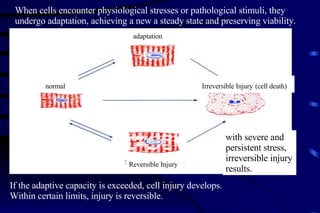
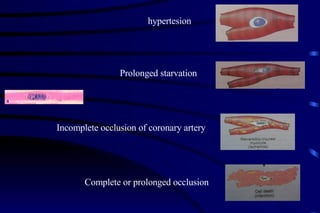
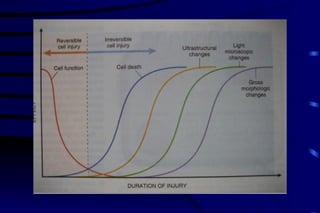
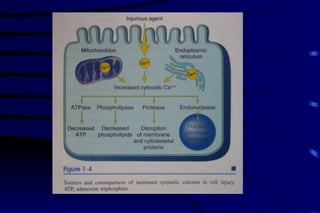
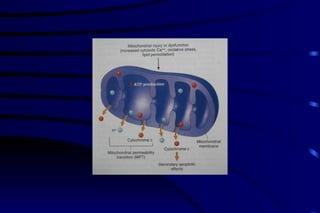
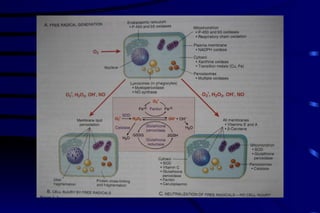
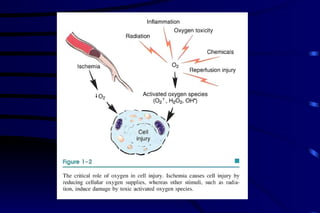
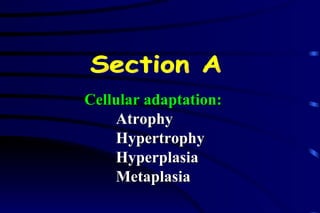
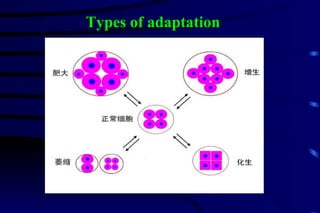
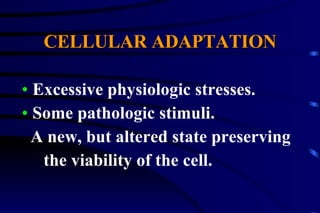
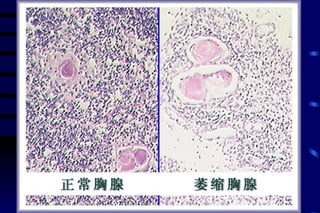
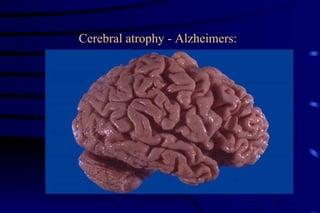
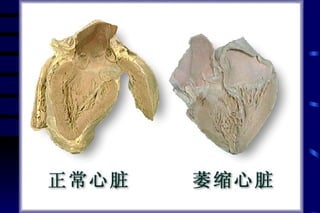
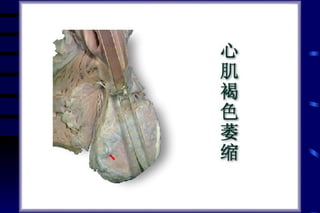
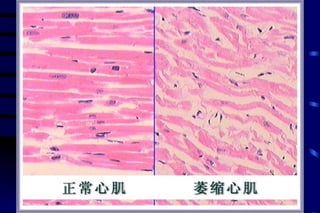
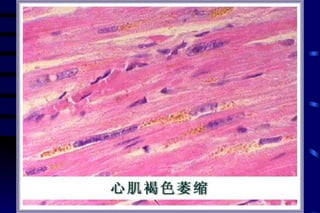
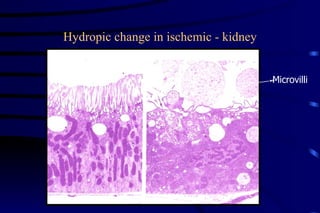
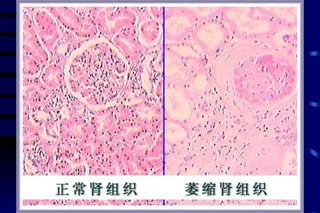
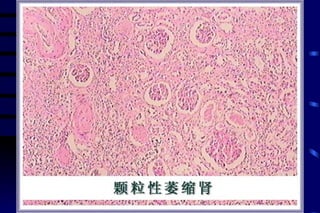
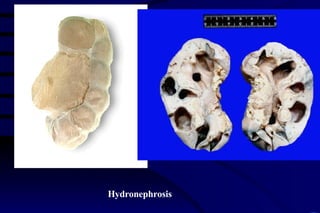
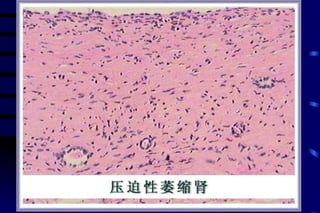
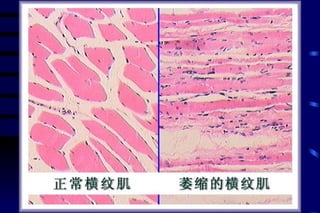
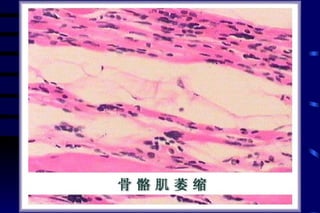
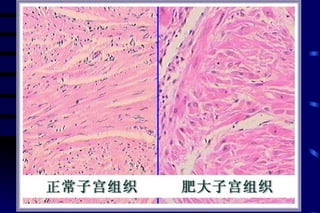
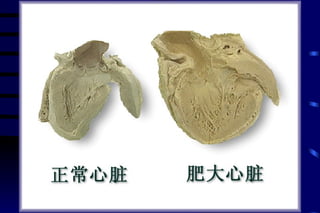
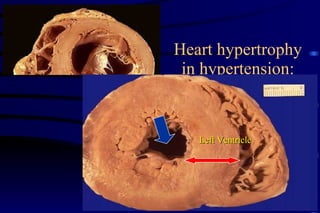
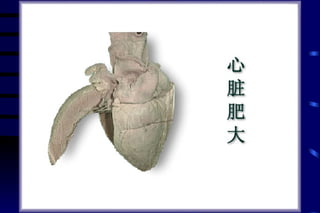
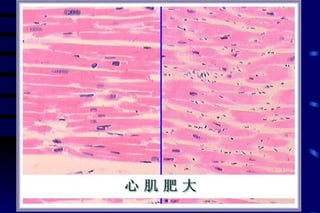
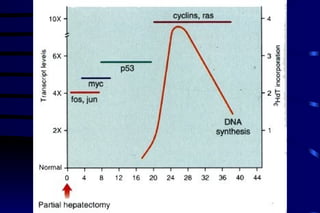
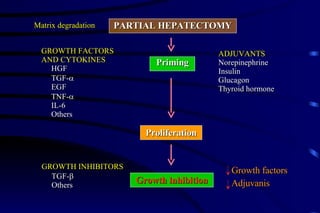
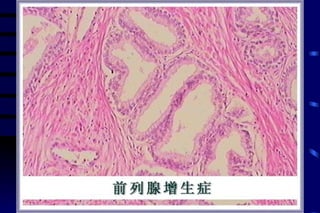
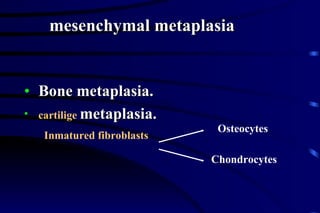
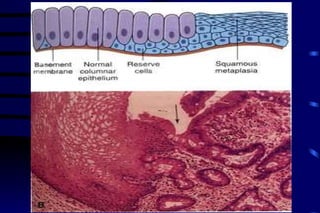
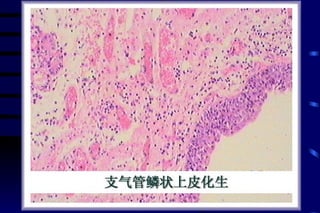
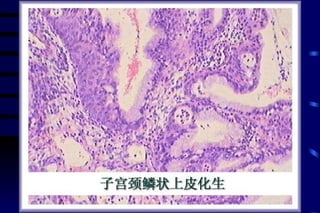
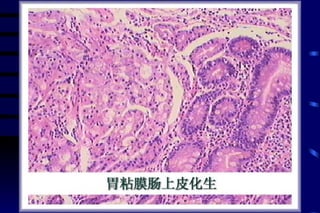
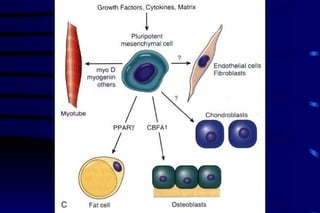
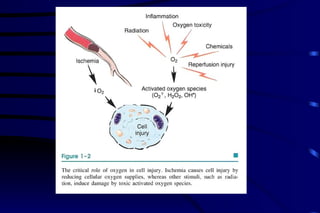
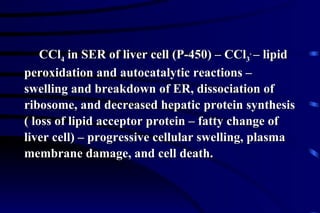
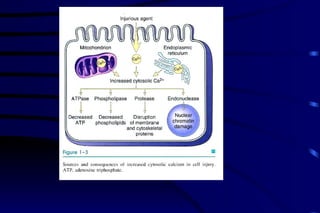
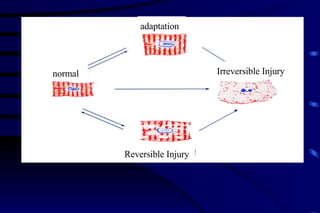
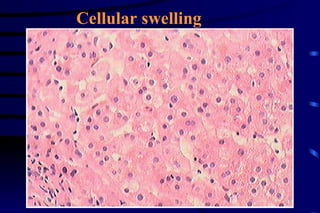
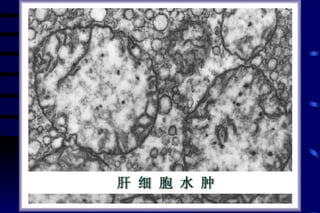
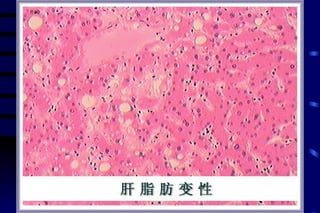
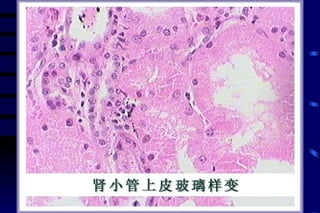
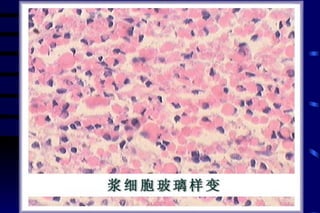
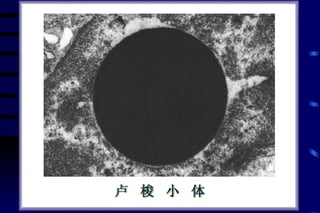
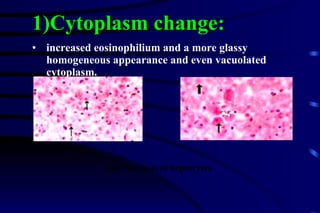
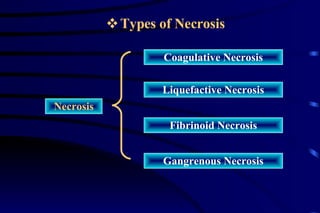
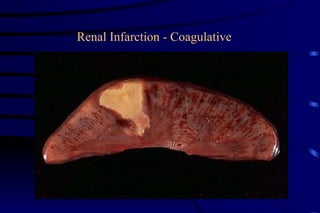
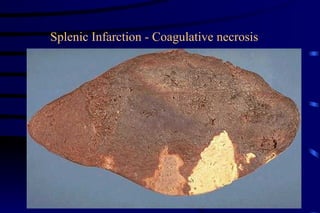
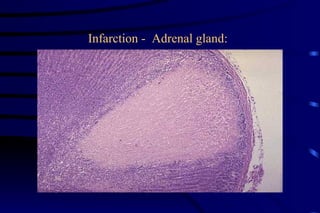
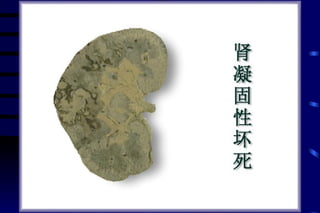
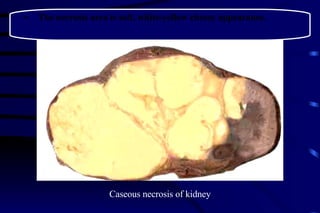
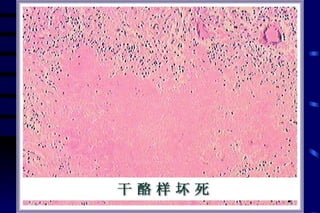
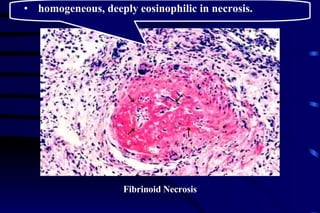
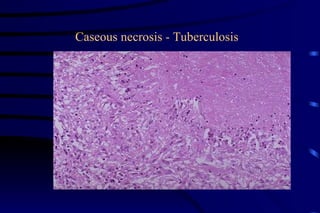
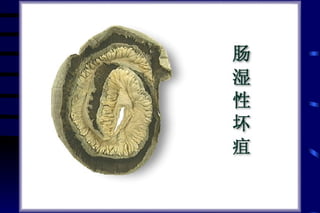
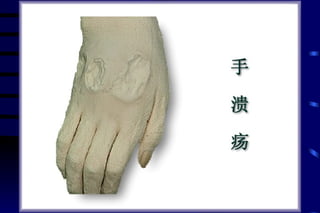
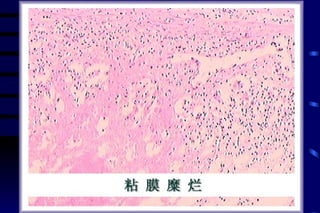
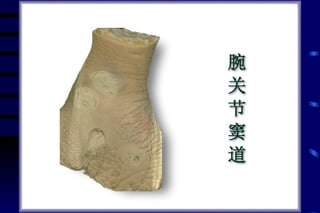
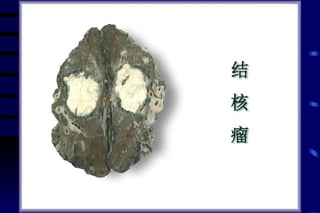
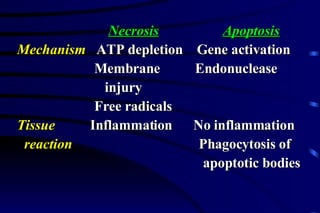
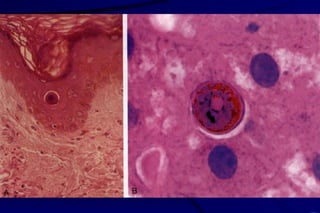
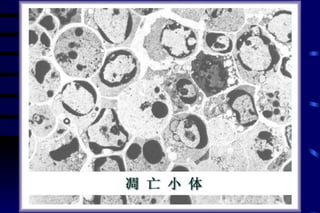
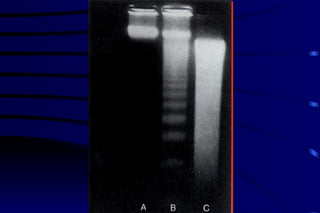
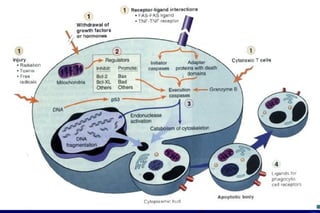
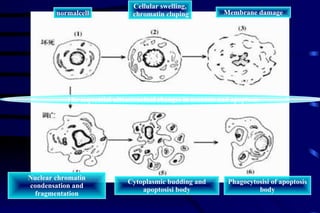
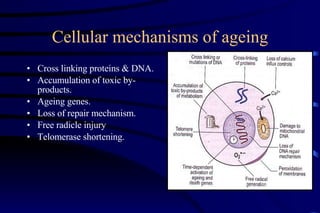
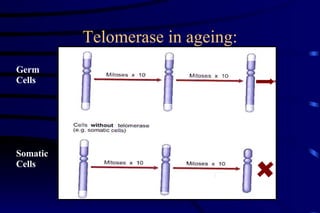
The document summarizes cellular adaptation and injury. It states that cells can undergo adaptation to achieve a new steady state and preserve viability when faced with physiological stresses, but severe or persistent stress can lead to reversible or irreversible injury. It describes different types of cellular adaptation like atrophy, hypertrophy, hyperplasia and metaplasia. It also discusses various causes of cellular injury like hypoxia, chemicals, infections and genetic factors. The key mechanisms of injury involve defects in membranes, ATP production and protein/DNA integrity.















































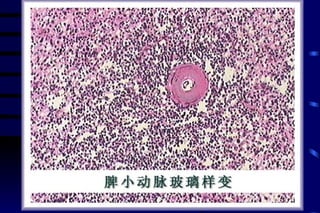
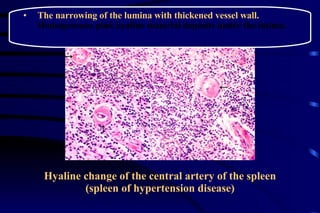

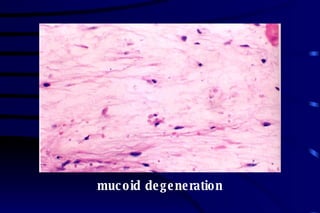

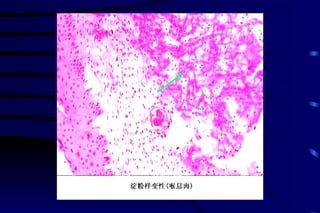







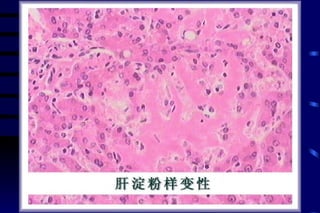

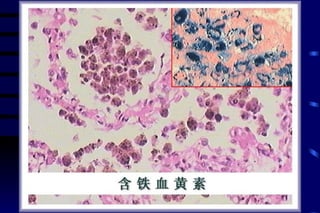
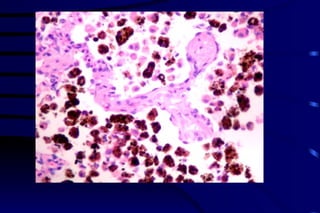
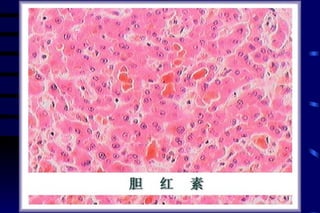

















































































![CELLULAR INJURY, ADAPTATION AND CELLULAR DEATH [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/cellularinjuryadaptationandcellulardeathautosaved-241015142117-5b666f59-thumbnail.jpg?width=640&height=640&fit=bounds)












































