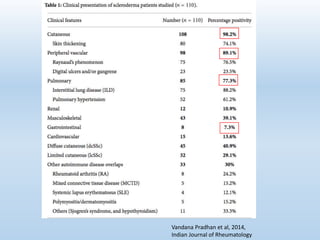
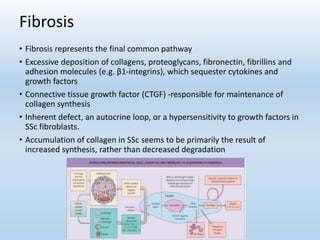
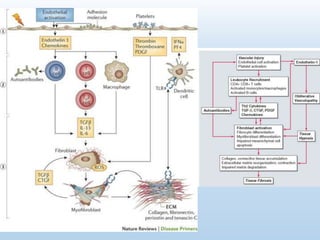
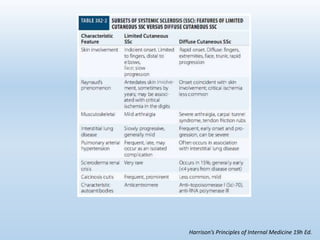
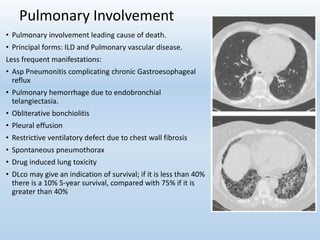
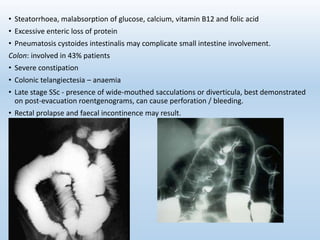
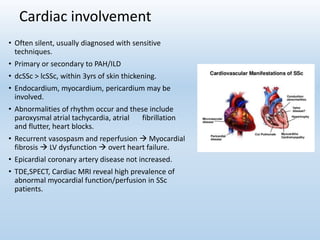
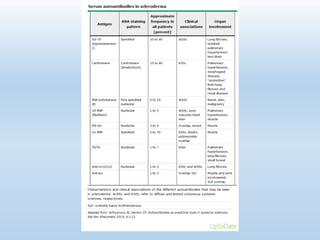
Scleroderma is an autoimmune connective tissue disease that causes hardening of the skin and internal organs. It is classified into limited and diffuse subtypes based on the extent of skin involvement. Raynaud's phenomenon, skin thickening, and pulmonary and gastrointestinal issues are common clinical manifestations. The underlying pathogenesis involves vascular dysfunction, immune dysregulation, and fibrosis. Management focuses on treating individual organ system complications. Prognosis depends on the specific organ systems affected and can range from relatively mild to severe with significant morbidity and mortality.






































![Pulmonary Artery Hypertension
• Advances in the treatment of pulmonary artery hypertension:
- Endothelin receptor antagonists (bosentan, sitaxsentan, ambrisentan)
- Phosphodiesterase type 5 inhibitors (sildenafil, tadalafil)
- Prostacyclin analogues iloprost [inhaled], epoprostenol [intravenous],
treprostinil [subcutaneous] are approved for pulmonary arterial hypertension
• Immunosuppressive agents: mycophenolate mofetil, azathioprine, chlorambucil,
5-fluorouracil, cyclosporine.
• Autologous hematopoietic stem cell transplantation is currently being studied.](https://image.slidesharecdn.com/systemicsclerosis1-180524074533/85/Systemic-Sclerosis-2017-53-320.jpg)































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)




