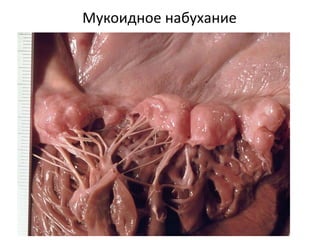
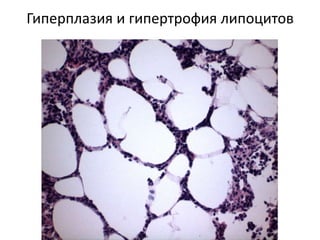
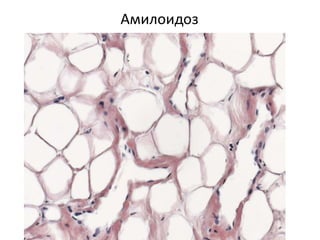
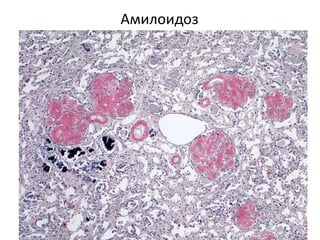
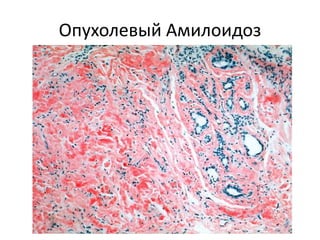
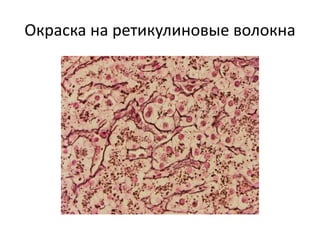
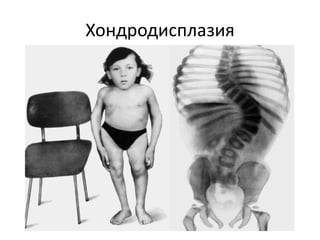
Документ обсуждает стромально-сосудистые дистрофии, которые включают белковые, жировые и амилоидозные дистрофии, возникающие вследствие нарушений обмена в соединительной ткани и кровеносных сосудах. Приводится классификация и описание различных форм стабильных и реактивных изменений, включая мукоидное и фибриноидное набухание, а также гиалиноз. Описаны этиологические факторы и патогенез различных форм заболеваний, включая амилоидоз, с акцентом на диагностические методы и клинические проявления.