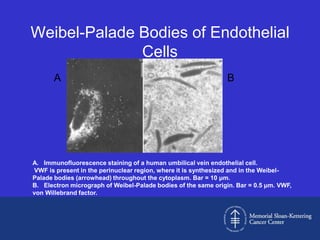
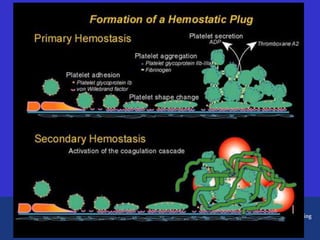
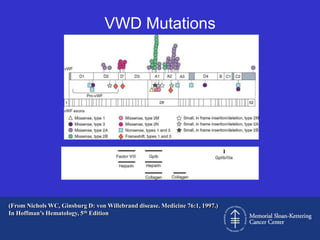
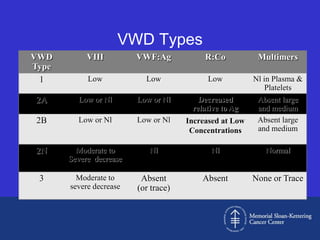
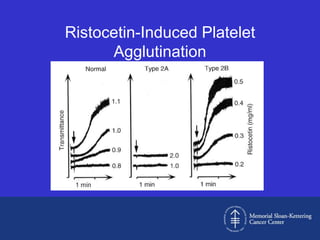
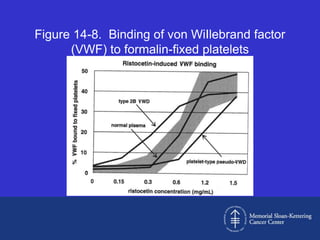
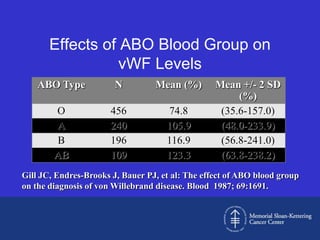
This document discusses three cases of von Willebrand disease (VWD) and provides information about VWD.
Case 1 is described as type 1 VWD based on test results showing reduced but present von Willebrand factor (vWF) antigen and activity. Case 2 is diagnosed as vWD Normandy based on low factor VIII and normal vWF antigen and multimers. Case 3 has complete deficiency of vWF and is diagnosed as type 3 VWD.
The document then provides details on the classification, symptoms, diagnosis, and treatment of VWD, the most common inherited bleeding disorder. It is caused by mutations in vWF resulting in deficient or defective vWF.