Download as PDF, PPTX














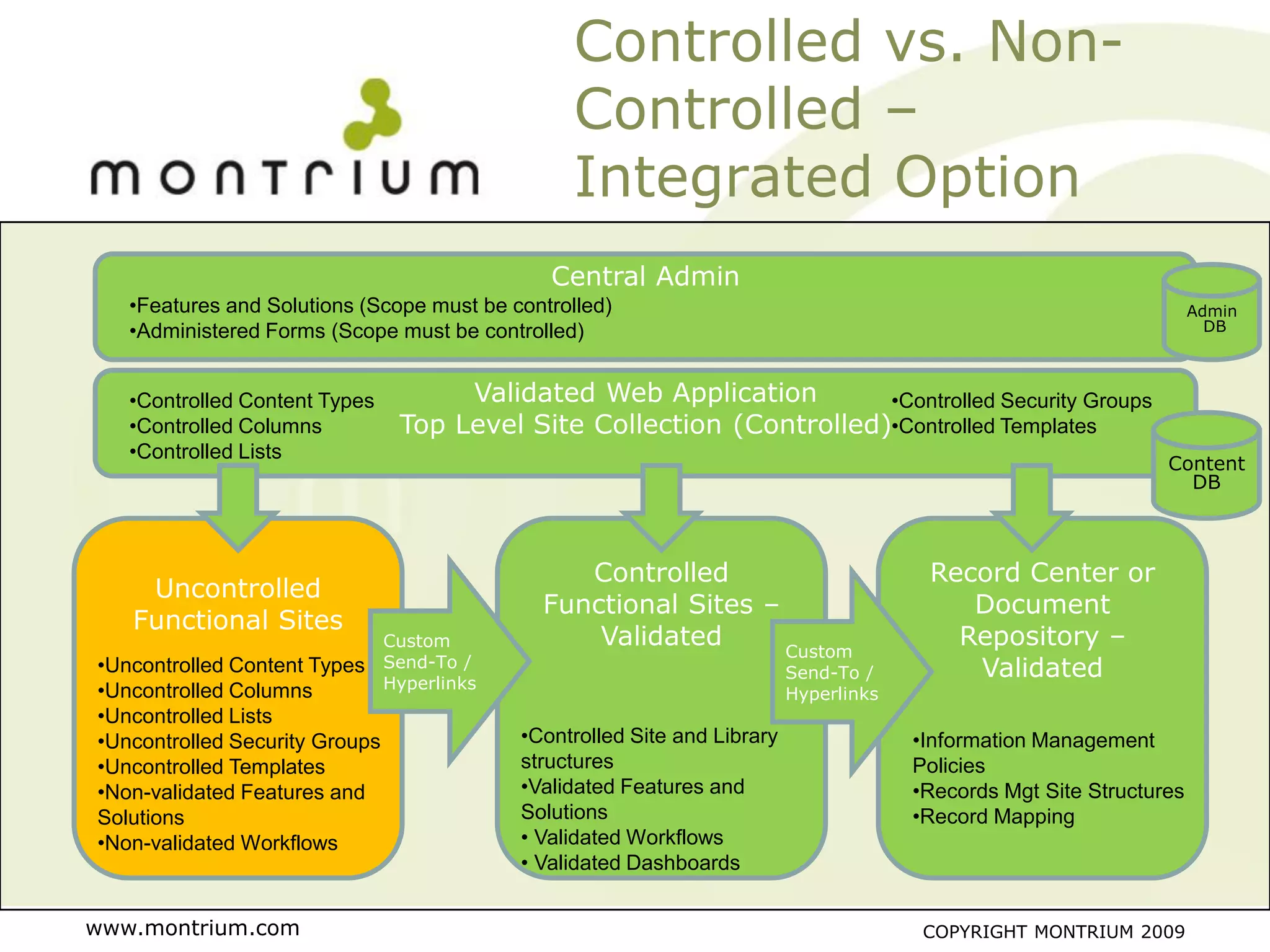
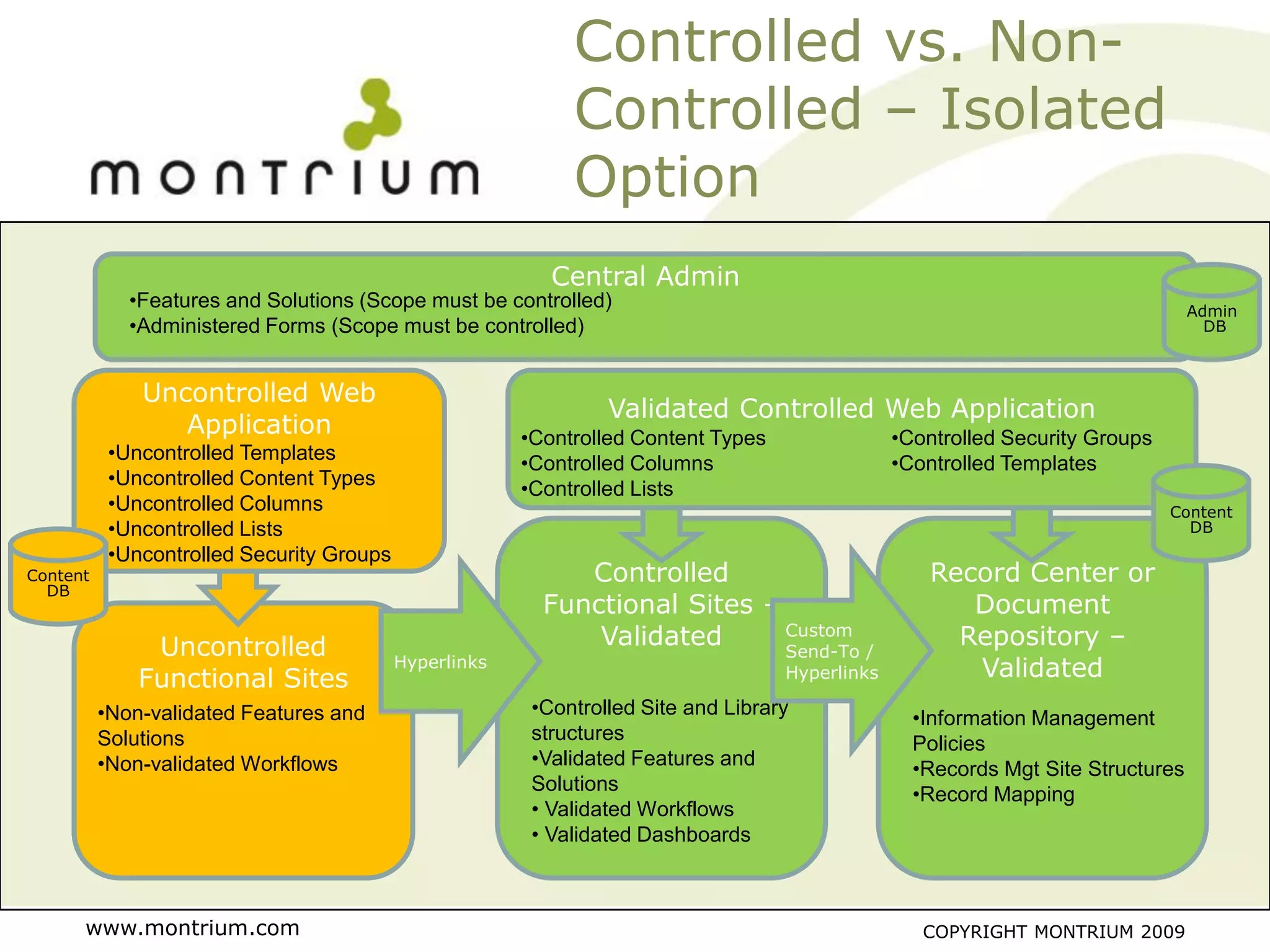
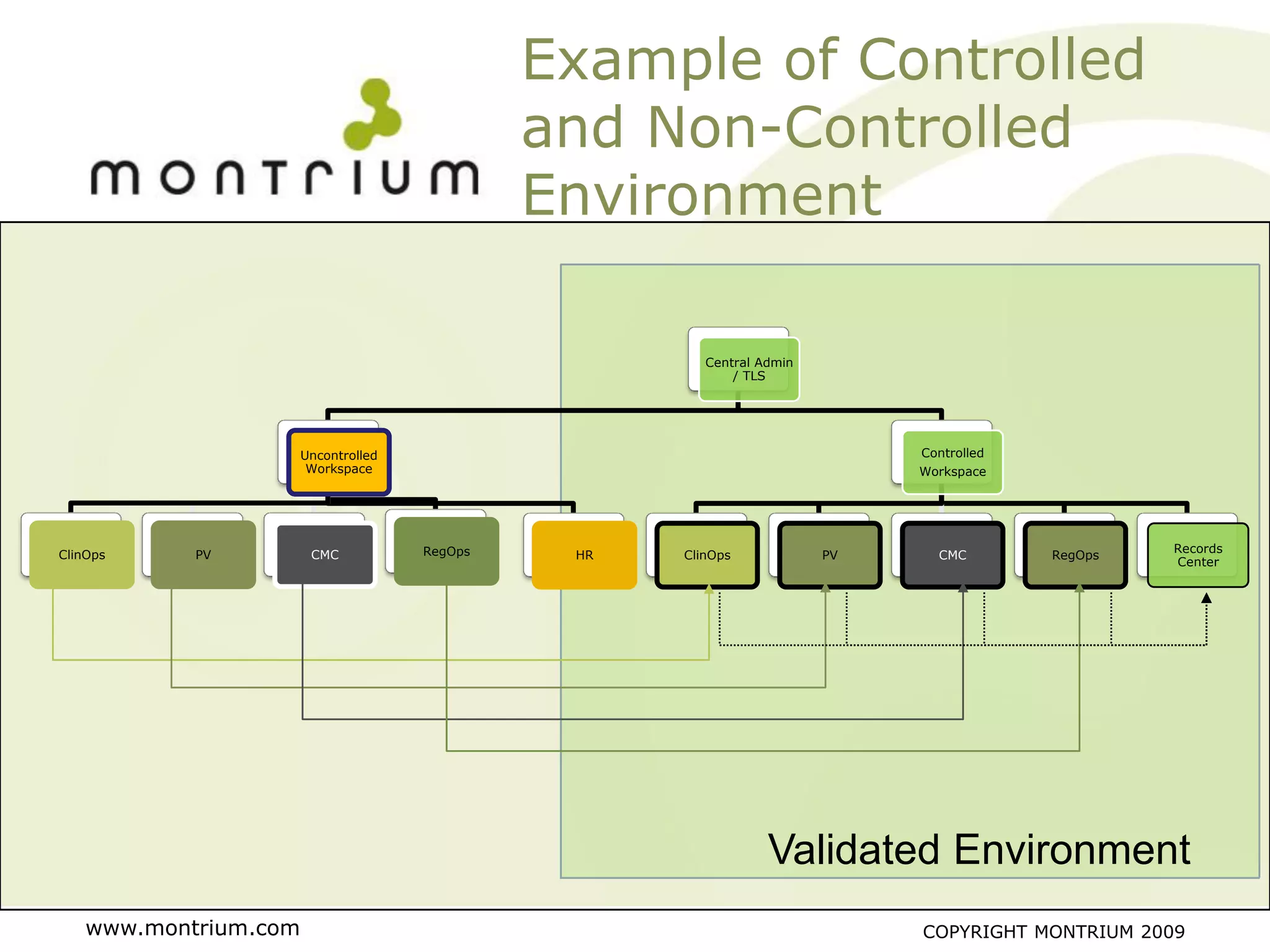






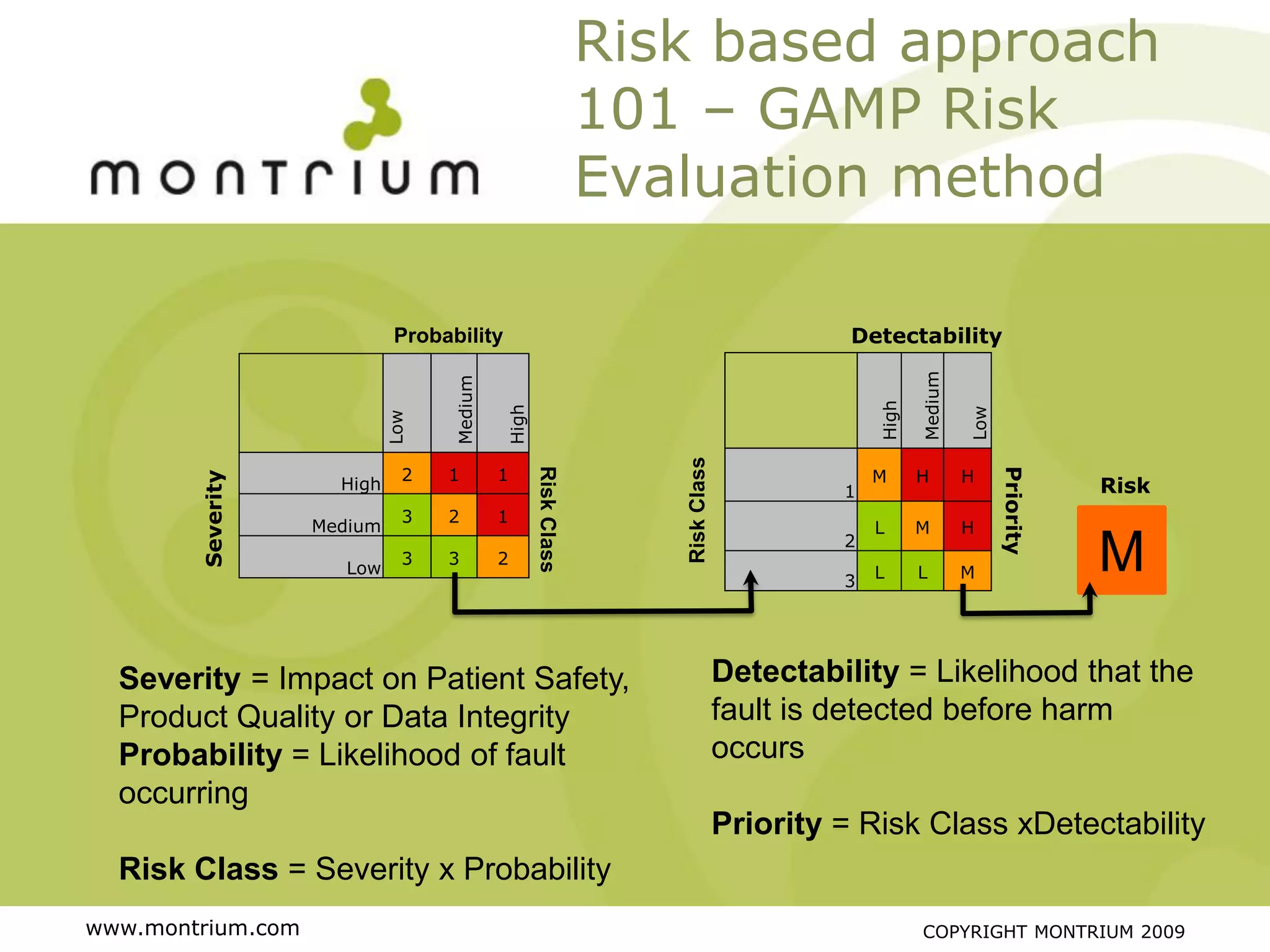

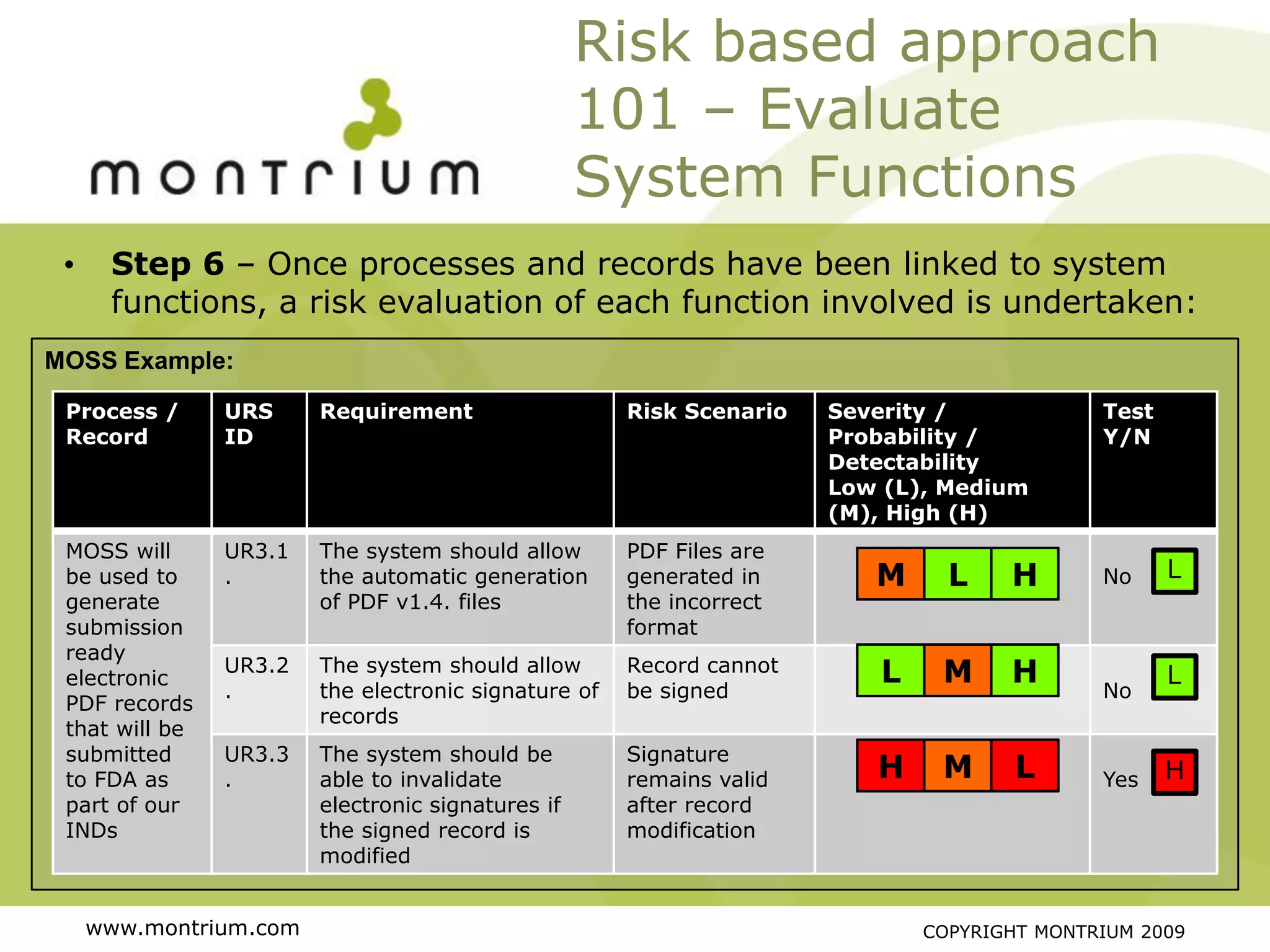


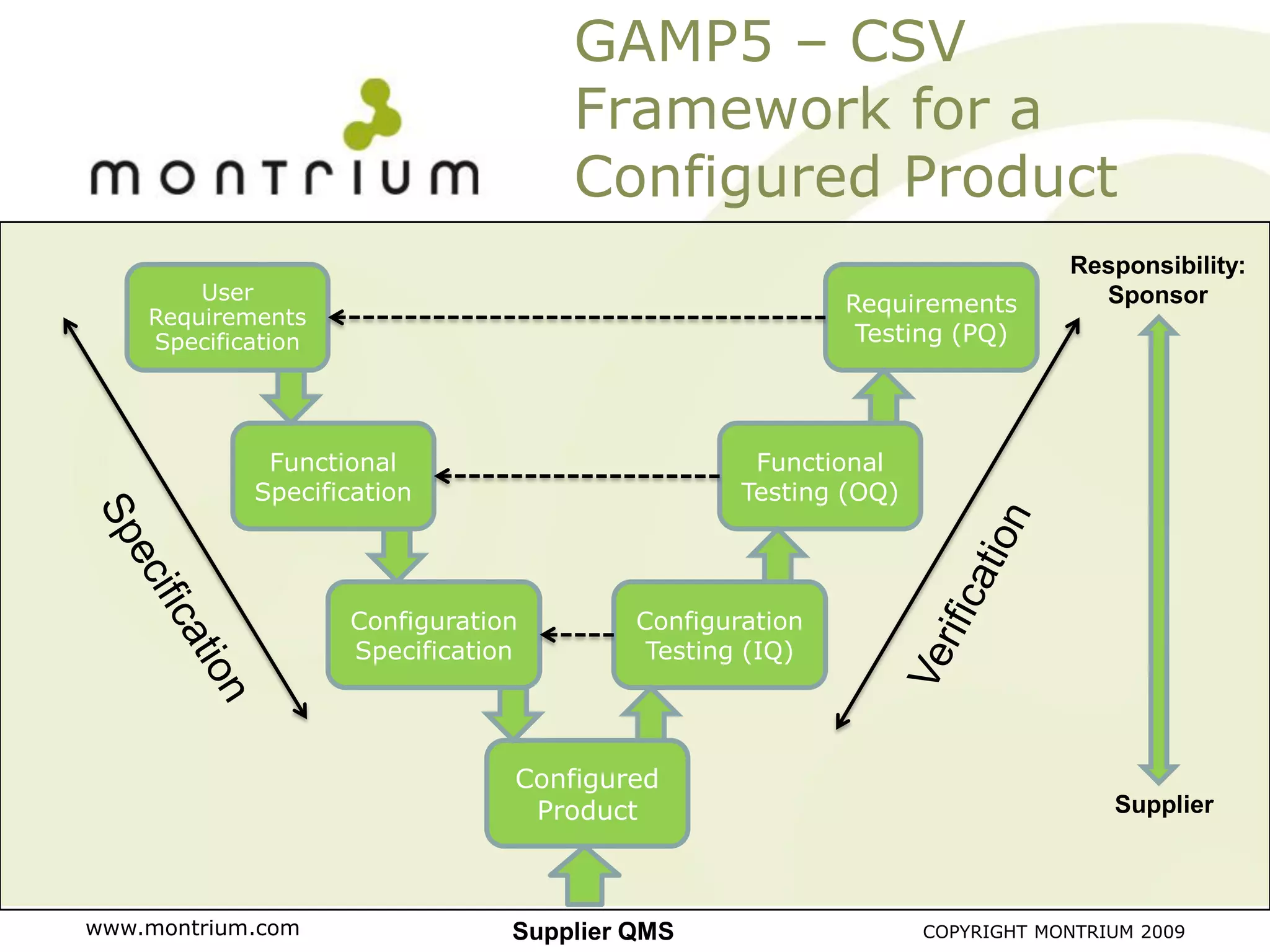
















The document outlines a webinar presented by Paul Fenton on utilizing SharePoint in compliance with 21 CFR Part 11 for the pharmaceutical industry, covering validation processes in GxP environments. Key topics include regulations, risk evaluation, and best practices for managing electronic records and configurations. It emphasizes a risk-based validation approach and the importance of maintaining controlled environments for regulated activities.











































































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)

















