Introduction
• Bioinformatics isan emerging field of science that deals with the
application of computers to the collection, organization, analysis,
manipulation, presentation, and sharing of biological data.
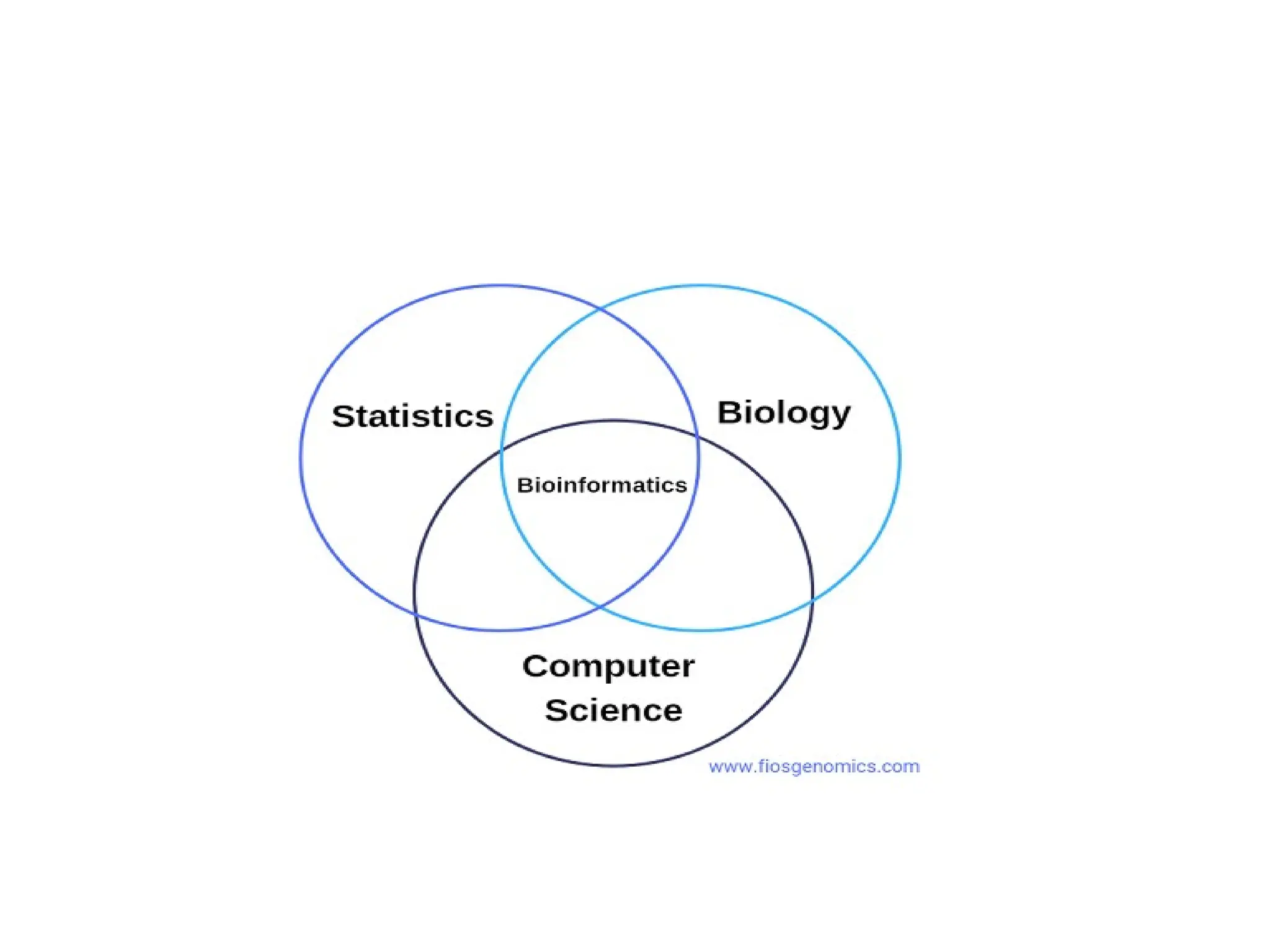
• Bioinformatics is an interdisciplinary field directly involving
molecular biology, genetics, computer science, mathematics, and
statistics.
• The central component of bioinformatics is the study of the best
ways to design and operate biologic databases.
• As a large amount of nucleotide and protein sequence data are
obtained via various research techniques, along with other types
of information stored in primary and secondary biological
databases, scientists started to use computers to obtain and
analyze biological data in their daily research with bioinformatics
tools.
4.
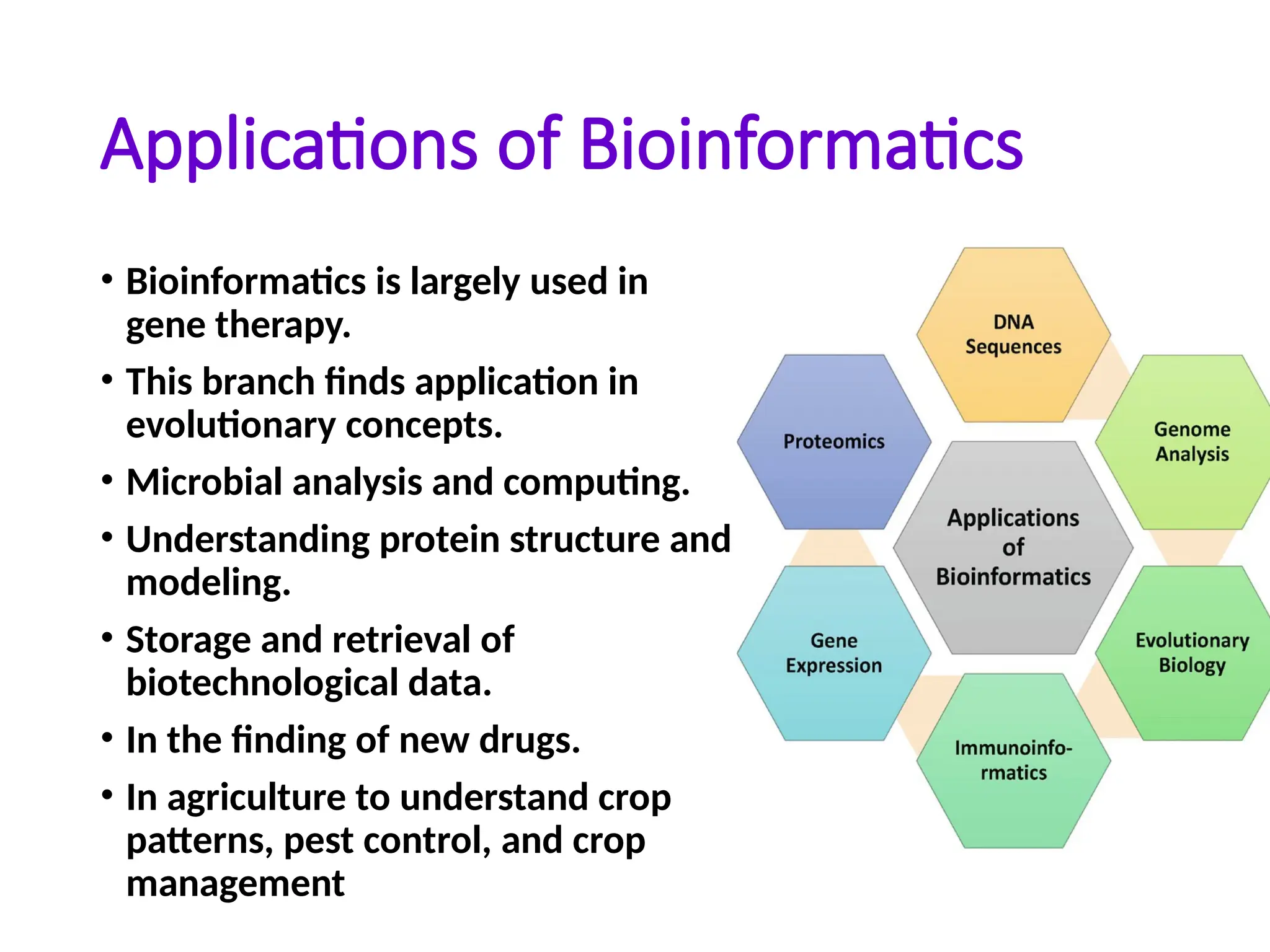
Applications of Bioinformatics
•Bioinformatics is largely used in
gene therapy.
• This branch finds application in
evolutionary concepts.
• Microbial analysis and computing.
• Understanding protein structure and
modeling.
• Storage and retrieval of
biotechnological data.
• In the finding of new drugs.
• In agriculture to understand crop
patterns, pest control, and crop
management
5.
Data bases
• Biologicaldata are complex, exception-ridden,
vast and incomplete. Therefore several databases
has been createdand interpreted to ensure
unambiguous results. Acollection of biological
data arranged in computer readable form that
enhances the speed of search andretrieval and
convenient to use is called biologicaldatabase. A
good database must have updated information.
6.
• A rangeof information like biological
sequences,structures, binding sites, metabolic
interactions, molecularaction, functional
relationships, protein families, motifs and
homologous can be retrieved by using biological
7.
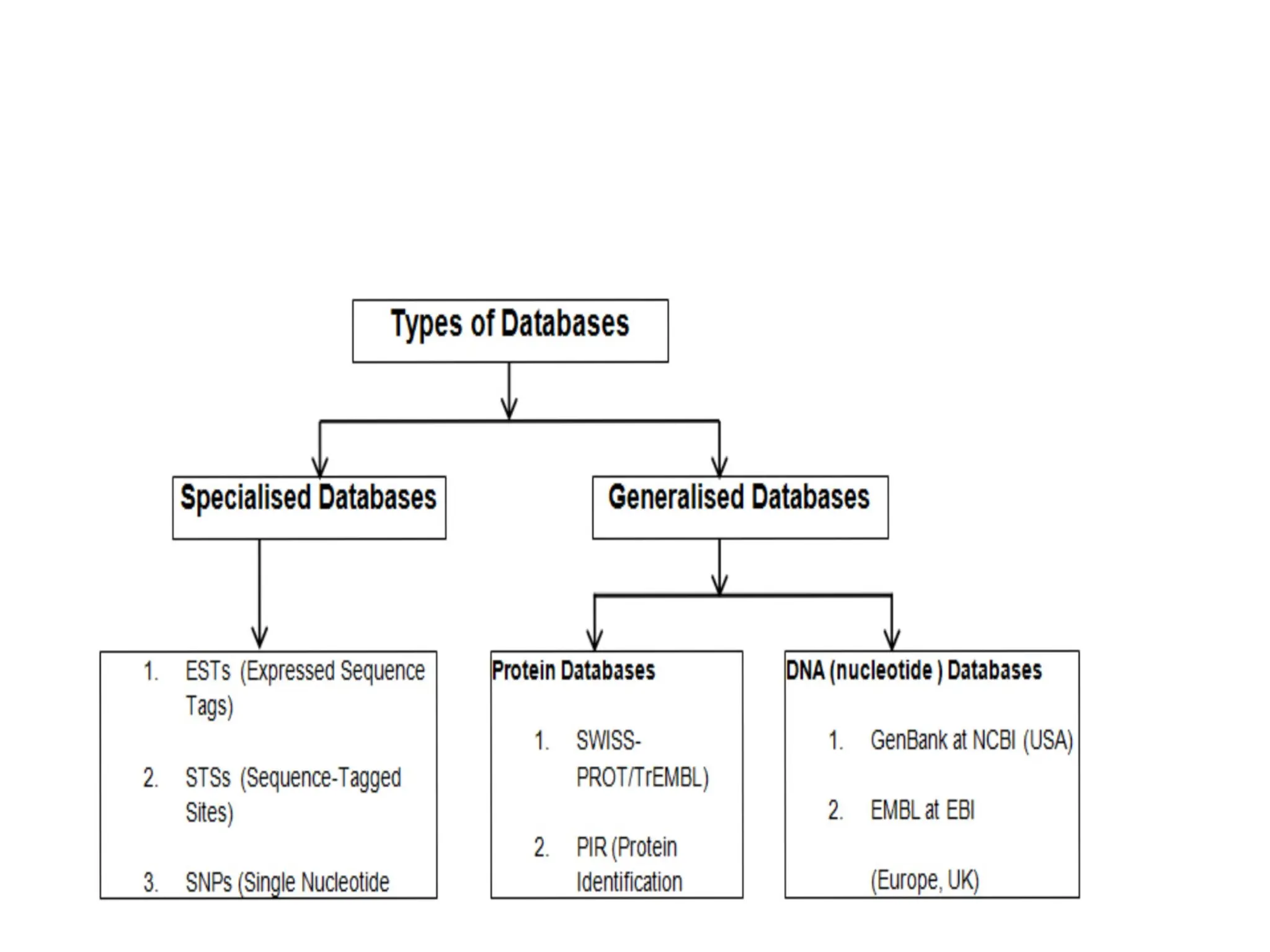
Types of databases
• There are basically 3 types of biological databases are as follows.
• 1. Primary databases:
• It can also be called an archival database since it archives the experimental
results submitted by the scientists. The primary database is populated with
experimentally derived data like genome sequence, macromolecular
structure, etc. The data entered here remains uncurated(no modifications are
performed over the data).
• It contains unique data obtained from the laboratory and these data are
made accessible to normal users without any change.
• The data are given accession numbers when they are entered into the
database. The same data can later be retrieved using the accession number.
Accession number identifies each data uniquely and it never changes.
• Examples –
• Examples of Primary database- Nucleic Acid Databases are GenBank and
DDBJ
• Protein Databases are PDB,SwissProt,PIR,TrEMBL,Metacyc, etc.
8.
• 2. SecondaryDatabase:
• The data stored in these types of databases are the analyzed
result of the primary database. Computational algorithms are
applied to the primary database and meaningful and
informative data is stored inside the secondary database.
• The data here are highly curated(processing the data before it is
presented in the database). A secondary database is better and
contains more valuable knowledge compared to the primary
database.
• Examples –
• Examples of Secondary databases are as follows.
• InterPro (protein families, motifs, and domains)
• UniProt Knowledgebase (sequence and functional information
on proteins)
9.
• 3. CompositeDatabases:
• The data entered in these types of databases are first
compared and then filtered based on desired criteria.
• The initial data are taken from the primary database, and
then they are merged together based on certain
conditions.
• It helps in searching sequences rapidly. Composite
Databases contain non-redundant data.
• Examples –
• Examples of Composite Databases are as follows.
• Composite Databases -OWL,NRD and Swissport +TREMBL
10.
• There arevarious types of databases to store
information about biological patterns of DNA.
Most of these databases contain evolving
information and therefore have gone through
several revisions, since they were first
introduced. With the advancement of
technology, a number of new software for
DNA computing has been developed in the
field of computer.
12.
Pubmed
• PubMed isa free resource developed and maintained by the
National Center for Biotechnology Information (NCBI), a
division of the U.S. National Library of Medicine (NLM), at the
National Institutes of Health (NIH).
• PubMed comprises over 22 million citations and abstracts for
biomedical literature indexed in NLM’s MEDLINE database, as
well as from other life science journals and online books.
PubMed citations and abstracts include the fields of
biomedicine and health, and cover portions of the life sciences,
behavioral sciences, chemical sciences, and bioengineering.
PubMed also provides access to additional relevant websites
and links to other NCBI resources, including its various
molecular biology databases.
• PubMed uses NCBI's Entrez search and retrieval system.
PubMed does not include the full text of the journal article;
however, the abstract display of PubMed citations may provide
links to the full text from other sources, such as directly from a
publisher’s website or PubMed Central (PMC).
13.
• PubMed wasfirst released in January 1996
as an experimental database under the
Entrez retrieval system with full access to
MEDLINE. The word "experimental" was
dropped from the website in April 1997, and
on June 26, 1997, free MEDLINE access via
PubMed was announced at a Capitol Hill
press conference. Use of PubMed has grown
exponentially since its introduction: PubMed
searches numbered approximately 2 million
• for the month of June 1997, while current
usage typically exceeds 3.5 million searches
per day.
• PubMed was significantly redesigned in
2000 to integrate new features such as
LinkOut,
• Limits, History, and Clipboard
14.
NCBI
• NCBI NationalCentre for Biotechnology Information
(http://www.ncbi.nlm.nih.gov/)
• Established as the government's response to the need for more
and better information processing methods to deal with vast
amount of data.
• Developed by National Library of Medicine (NLM) at the National
Institutes of Health (NIH) A comprehensive website for
biologists including: biology-related databases, tools for
viewing and analyzing automated systems for storing and
retrieval
• 3. NCBI along with EBI and CIB together form International
Sequence Database Collaboration which act as the chief working
unit and Information Centre. NCBI has 3 collaborative databases:
GenBank European Molecular Biology Laboratory (EMBL)
Database DNA Database of Japan (DDBJ)
16.
EMBL
• European BioinformaticsInstitute (EBI) is a center for research and
services in bioinformatics, and is part of European Molecular
Biology Laboratory(EMBL) • EMBL-EBI grew out of EMBL’s
pioneering work to provide public biological database to research
community.It is located on the Welcome Trust Genome Campus in
Hinxton, UK along with welcome trust sanger institute.•
• EMBL-EBI maintains a comprehensive range of freely available and
up-to- date databases, which collectively cover the full range of
molecular biology, from nucleotide sequences to full systems.
• • It has a mandate to make its tools and infrastructure freely
available to the global scientific community. • EMBL-EBI is a central
partner in global efforts to exchange information, set standards,
develop new methods, and curate complex information. • This
study includes exploration of the costs and cost savings involved in
using EMBL-EBI data and services, its value to users, and its impacts
on the wider commercial, healthcare, and research communities
17.
ExPasy
• The ExPASy(the Expert Protein Analysis System) World
Wide Web server (http://www.expasy.org), is provided as
a service to the life science community by a
multidisciplinary team at the Swiss Institute of
Bioinformatics (SIB). It provides access to a variety of
databases and analytical tools dedicated to proteins and
proteomics. ExPASy databases include SWISS-PROT and
TrEMBL, SWISS-2DPAGE, PROSITE, ENZYME and the SWISS-
MODEL repository. Analysis tools are available for specific
tasks relevant to proteomics, similarity searches, pattern
and profile searches, post-translational modification
prediction, topology prediction, primary, secondary and
tertiary structure analysis and sequence alignment. These
databases and tools are tightly interlinked
18.
• SWISS-2DPAGE
(http://www.expasy.org/ch2d/)----------- isa
database of proteins identified on two-
dimensionalpolyacrylamide of proteins
2DPAGEcontains data from a variety of human and
mouse Escherichia coli,Saccharomyces cerevisiae
and Dictyostelium discoideum.
• PROSITE (http://www.expasy.org/prosite/) is a
databaseof protein domains and families.PROSITE
contains biologically significant sites, patterns
andprofiles that help to reliably identify to which
known proteinfamily a new sequence belongs.
19.
BLAST
• With theincrease in DNA and
protein sequence databases, there is
a growing need for more faster and
efficient methods to analyze this
large amount of data. One of the
most commonly used bioinformatics
tools today to study DNA and
protein sequences is called BLAST.
• BLAST stands for Basic Local
Alignment Search Tool. It is a widely
used bioinformatics program that
was first introduced by Stephen
Altschul et al. in 1990 and has since
become one of the most popular
tools for sequence similarity search.
20.
Types of BLAST
•There are five types (variants) of BLAST that are differentiated based
on the type of sequence (DNA or protein) of the query and database
sequences.
• BLASTn:compares a nucleotide query sequence to a nucleotide
sequence database.
• BLASTp:compares a protein query sequence to a protein sequence
database.
• BLASTxcompares a nucleotide query sequence to a protein sequence
database by translating the query sequence into its six possible
reading frames and aligning them with the protein sequences.
• tBLASTn compares a protein query sequence to a nucleotide sequence
database by translating the nucleotide sequences in all six reading
frames and aligning them with the protein sequence.
• tBLASTx:compares a nucleotide query sequence to a nucleotide
sequence database by translating the query sequence in all six
reading frames and aligning them with the nucleotide sequences.
21.
Steps in BLAST
•BLAST works by comparing a query sequence to a
database of sequences to find regions of similarity. It
uses a heuristic approach to search for similarities in
the database, making it faster and more efficient.
• BLAST performs sequence alignment through the
following steps.
• Step 1: The first step is to create a lookup table or list
of words from the query sequence. This step is also
called seeding. First, BLAST takes the query sequence
and breaks it into short segments called words. For
protein sequences, each word is usually three amino
acids long, and for DNA sequences, each word is
usually eleven nucleotides long.
23.
• Step 2:The second step is to search a database of
known sequences to find any sequences that
contain the same words as the query sequence.
This is done to identify database sequences
containing the matching words.
24.
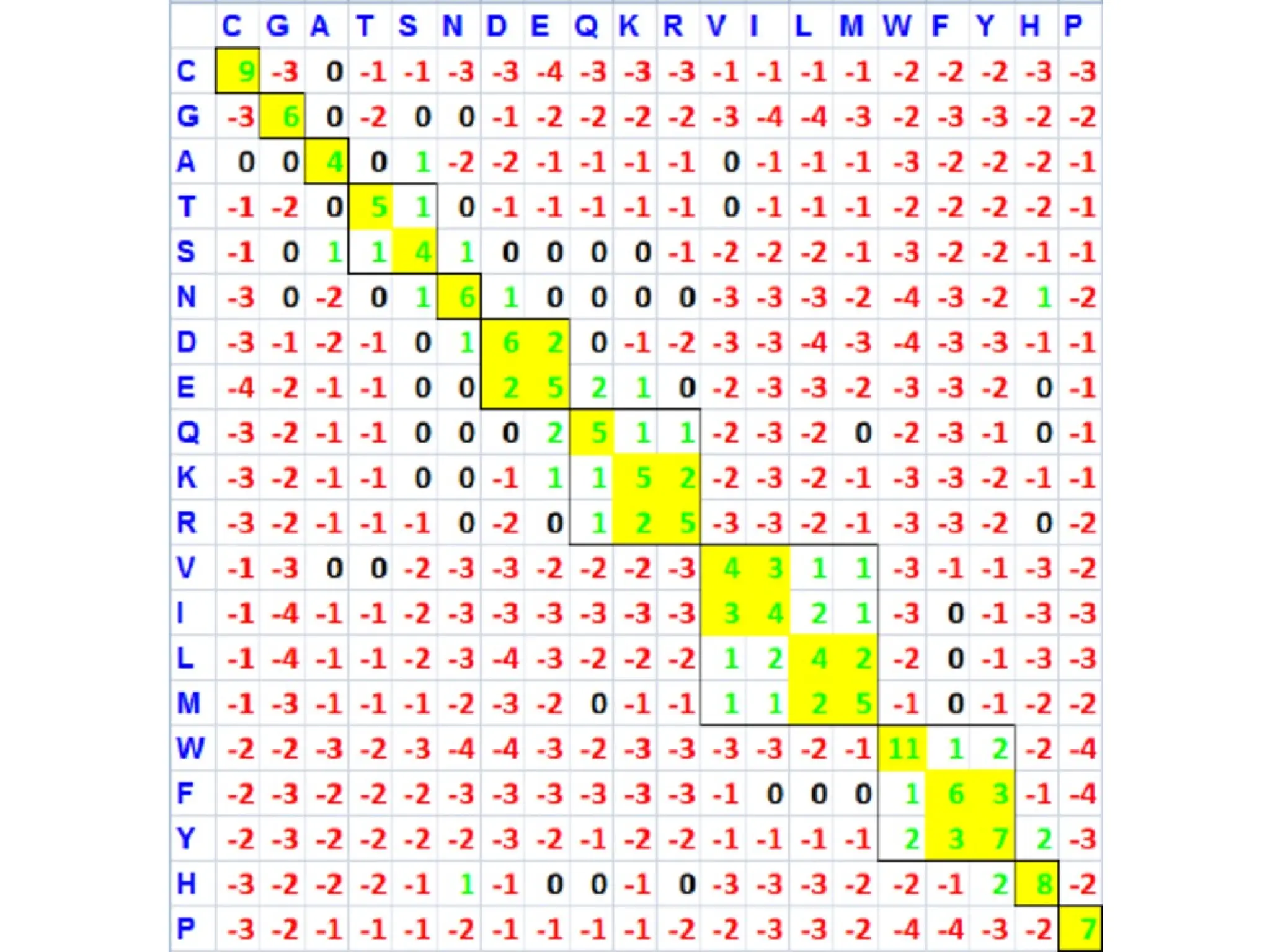
• Step 3:BLAST then scores the similarity of the matching words.
The matching of the words is scored by a given substitution
matrix. If a word is above a certain threshold, it is considered a
match.
• Two commonly used substitution matrices for protein
sequences are PAM (Percent Accepted Mutations) and BLOSUM
(Blocks Substitution Matrix). For nucleotide sequences, the
scoring matrix is based on match-mismatch scoring.
• Step 4: The fourth step involves pairwise alignment by
extending the words in both directions while counting the
alignment score using the same substitution matrix. If the score
drops below a certain threshold due to differences in the
sequences or mismatches, the alignment stops. The resulting
aligned segment pair without gaps is called the high-scoring
segment pair (HSP).
26.
Applications of BLAST
•BLAST has a wide range of applications. Some of the
most common applications are:
• BLAST can be used to identify unknown sequences by
comparing them with known sequences in a
database which helps in predicting the functions of
proteins or genes.
• BLAST can also be used in phylogenetic analysis
which is important for understanding the
evolutionary relationships between different species.
• BLAST can also be used to identify functionally
conserved domains within proteins which is
important for predicting the functions of proteins