Downloaded 186 times










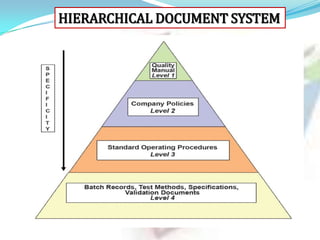















































The document provides information on documentation in the pharmaceutical industry including: 1. It emphasizes the importance of documentation for industries and describes protocols regarding documentation and their management. 2. Documentation is the key to GMP compliance and ensures traceability of all development, manufacturing, and testing activities. It provides a route for auditors to assess quality operations. 3. The most common types of documents used for GMP are described, including quality manuals, standard operating procedures, batch records, test methods, and specifications.
















































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)






































