Downloaded 15 times







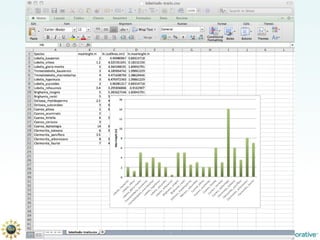

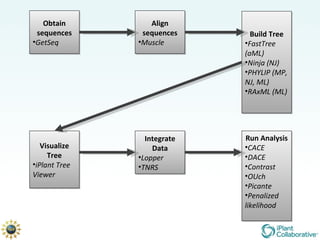
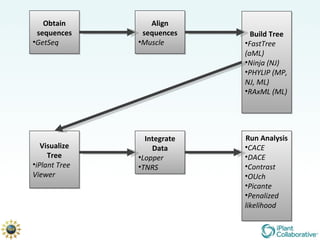



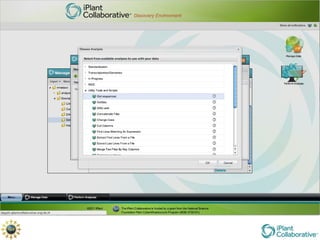
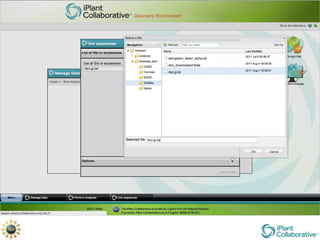
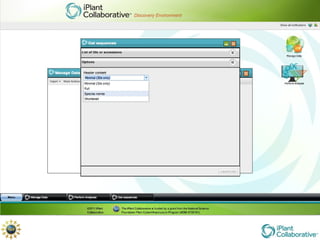
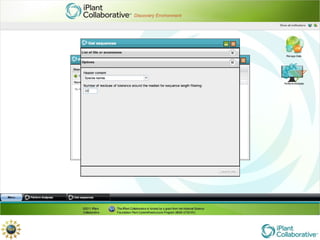
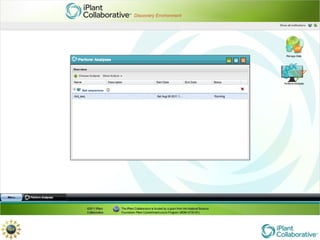
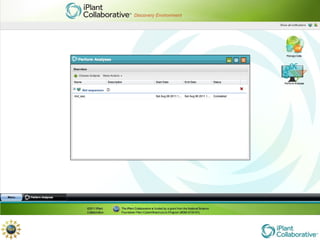
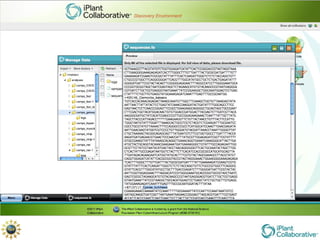
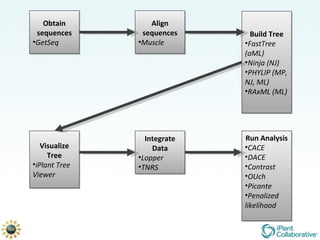

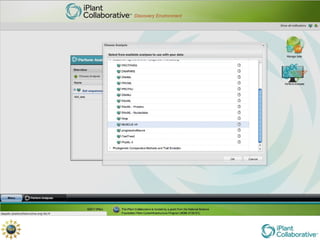
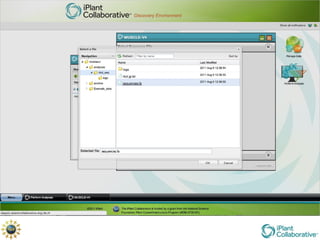
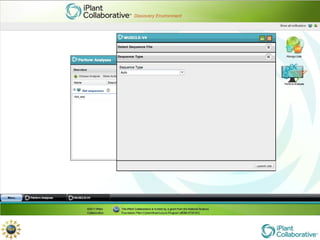
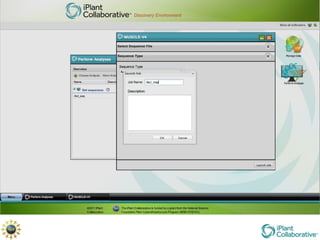
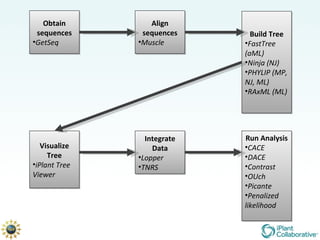


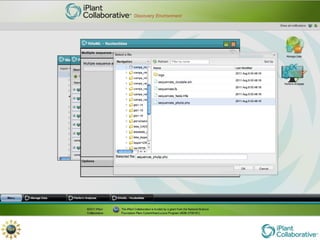
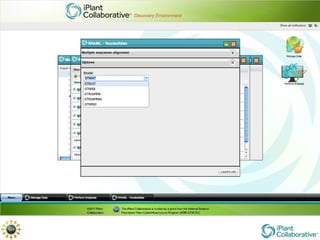
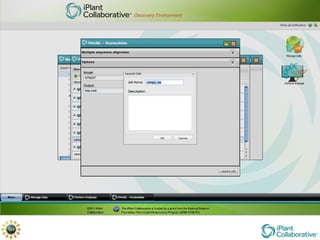
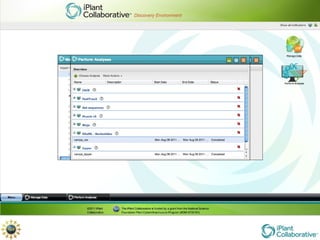
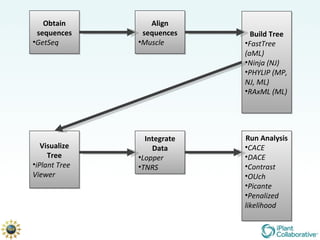

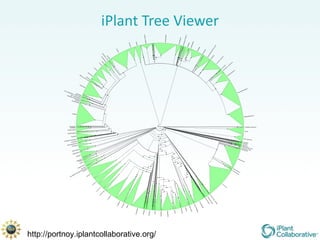
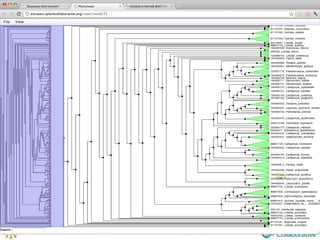
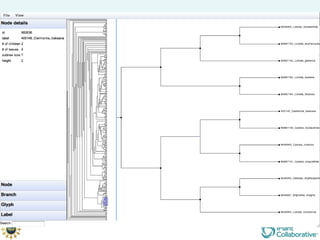
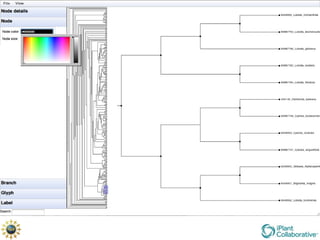
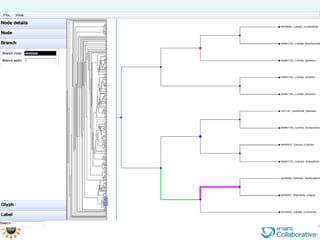
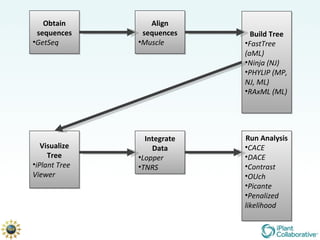


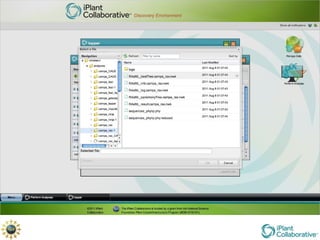
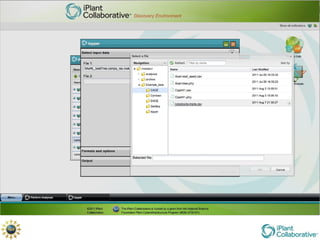
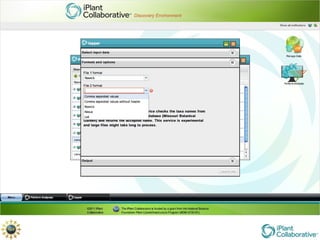
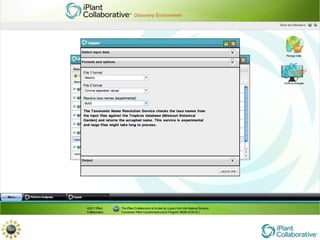
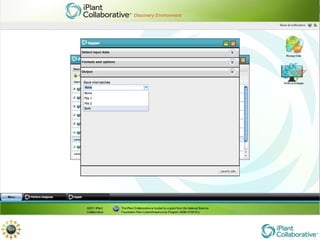
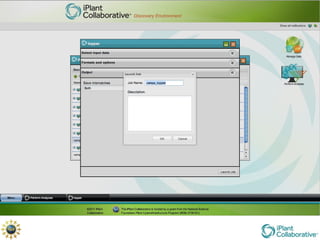
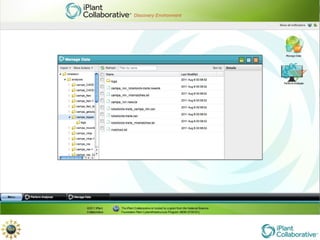
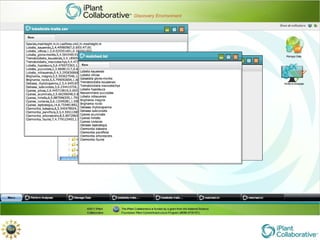
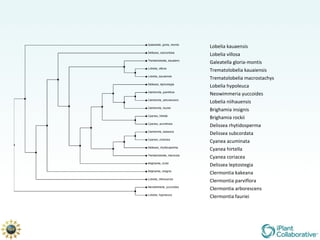

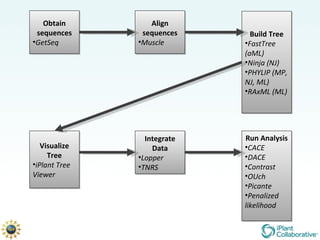

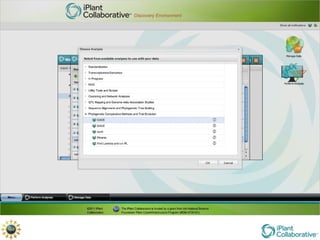
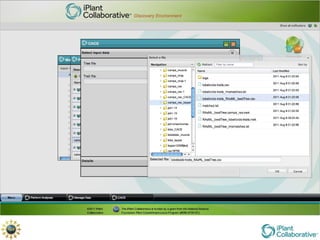
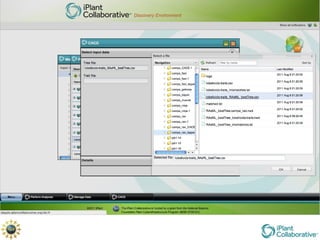

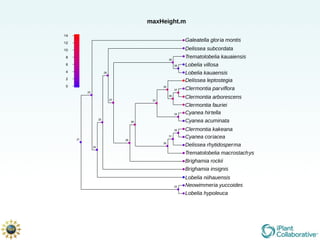
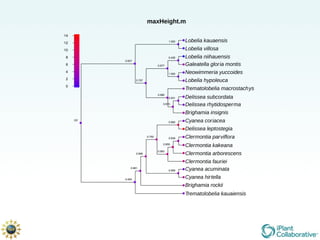

The document discusses the importance of phylogenetic workflows in building and analyzing the tree of life, emphasizing their significance in understanding evolutionary relationships in plant biology. It outlines various methods and tools for data acquisition, sequence alignment, tree building, and visualization, highlighting advancements in scalability and performance for handling large datasets. Additionally, it introduces specific applications and services like the iPlant Tree Viewer and Taxonomic Name Resolution Service (TNRS) to enhance data integration and analysis.

























































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)








