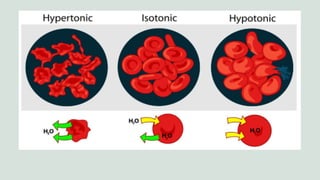
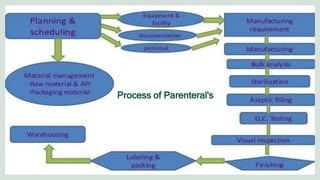
The document outlines the formulation, preparation, and evaluation of parenteral products, emphasizing their definitions, advantages, limitations, and classification. It details preformulation factors, methods of preparation, and the roles of various excipients, highlighting the importance of maintaining sterility and isotonicity during manufacturing. Various types of parenteral products are discussed, including small volume and large volume parenterals, infusions, and injectable emulsions, along with their corresponding routes of administration.










![ Particle Size and Shape - Study of particle size give information about solubility, dissolution
rate and absorption etc. These characteristics are determined by scanning electron microscope or
an optical microscope with polarizing attachments.
Ionization constant - This property is used to determine the PH-dependent solubility of a
compound. Potentiometric PH titration or PH-solubility analysis is used for determining the PKa
value. Ionization constant of a compound also helps in determining the degree of ionization of an
acid or base. Degree of ionization depends upon the PH.
For acidic drugs Pka ranges from 3-7.5 and for basic drugs Pka ranges from 7- 11.
Partition Coefficient (P) - It is a ratio of equilibrium concentration of drug in aqueous and
oily phases in contact with each other at a constant temperature.
Partition coefficient can be expressed as “P” = [Coil / Cwater],
Where, Coil -Organic phase concentration
Cwater - Aqueous phase concentration](https://image.slidesharecdn.com/parenteralsop-240424111123-f8b49ad2/85/Parenterals-from-Preparation-to-Evaluation-10-320.jpg)







































































![Industrial phar wps office [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/industrialphar-wpsofficeautosaved-201115070603-thumbnail.jpg?width=640&height=640&fit=bounds)



































