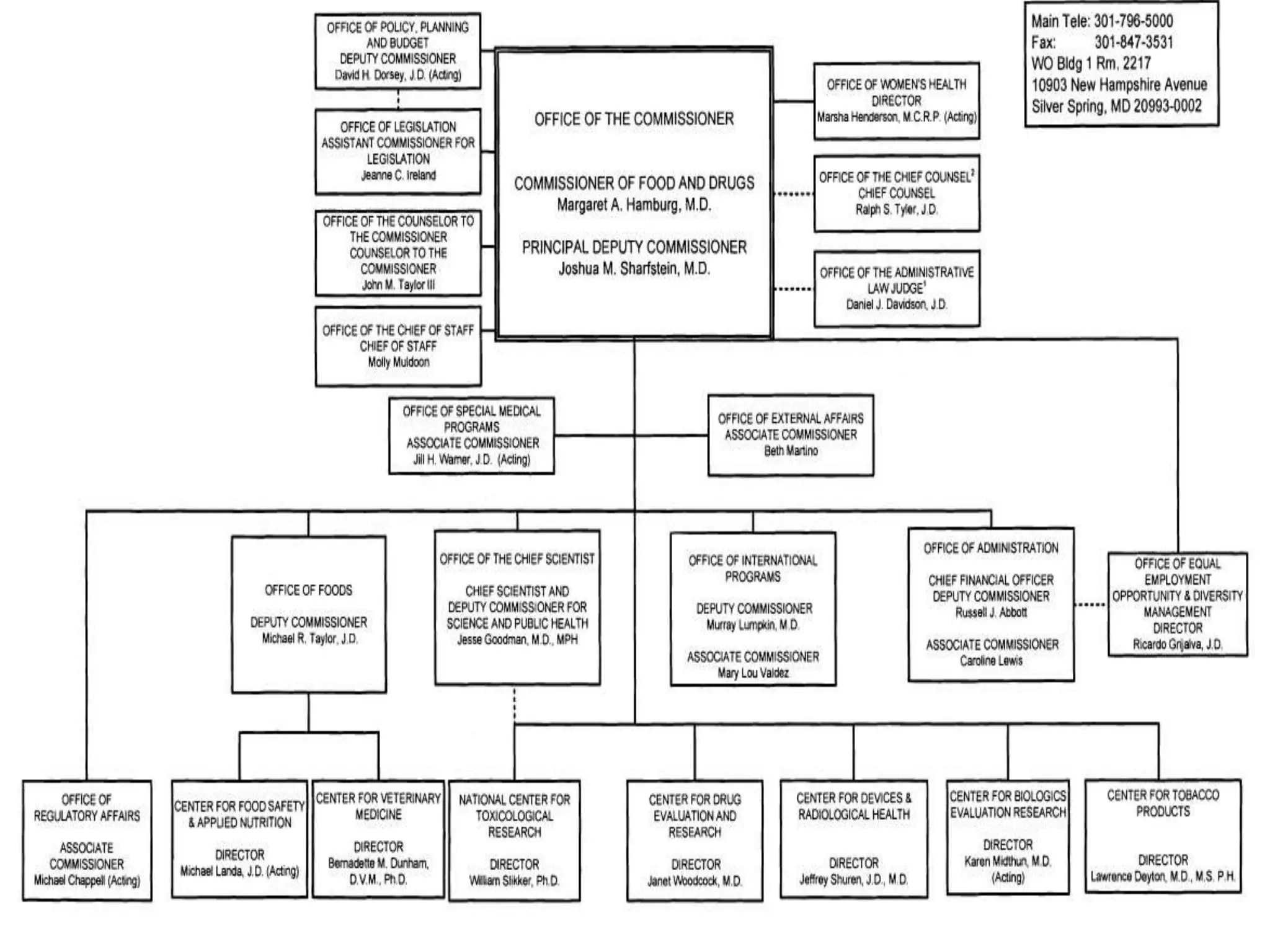
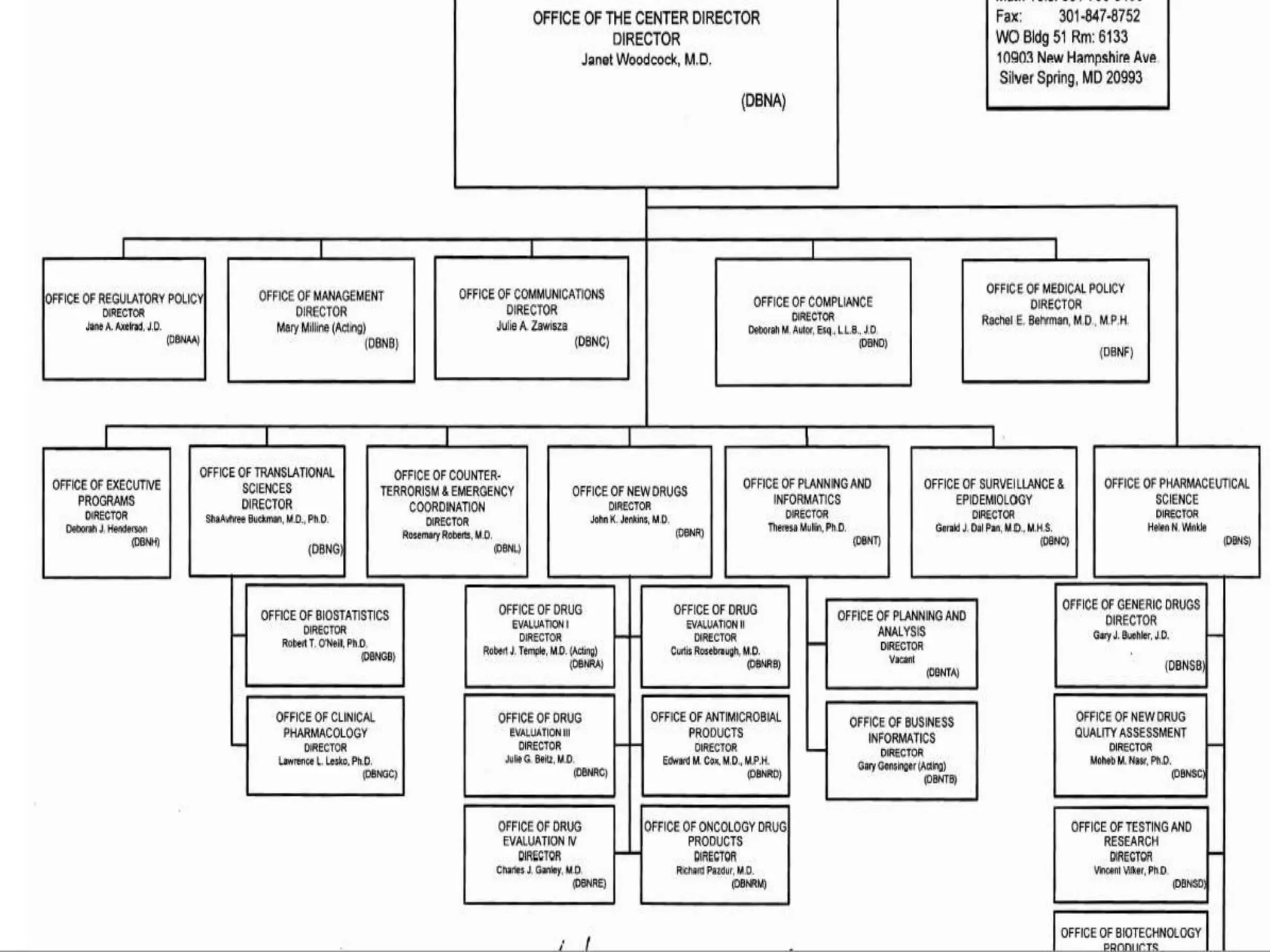
The document discusses the process for product registration and drug approval in the United States. All new drug products must complete a registration process with the FDA to ensure safety and efficacy. This involves submitting a New Drug Application (NDA) containing clinical and manufacturing data and information. The NDA goes through a review process by FDA teams with expertise in technical areas. The review determines if the drug is approved for marketing in the United States.