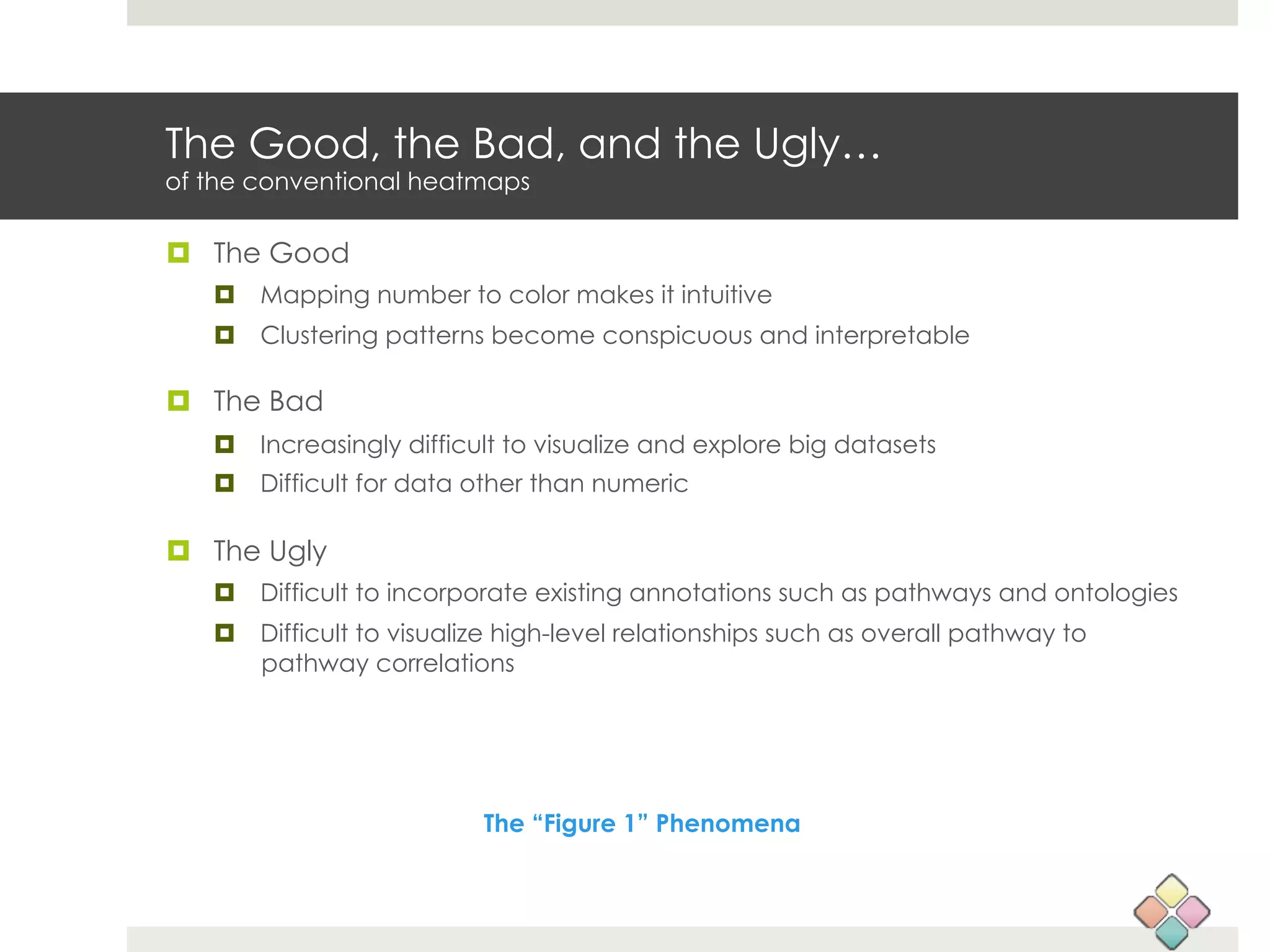
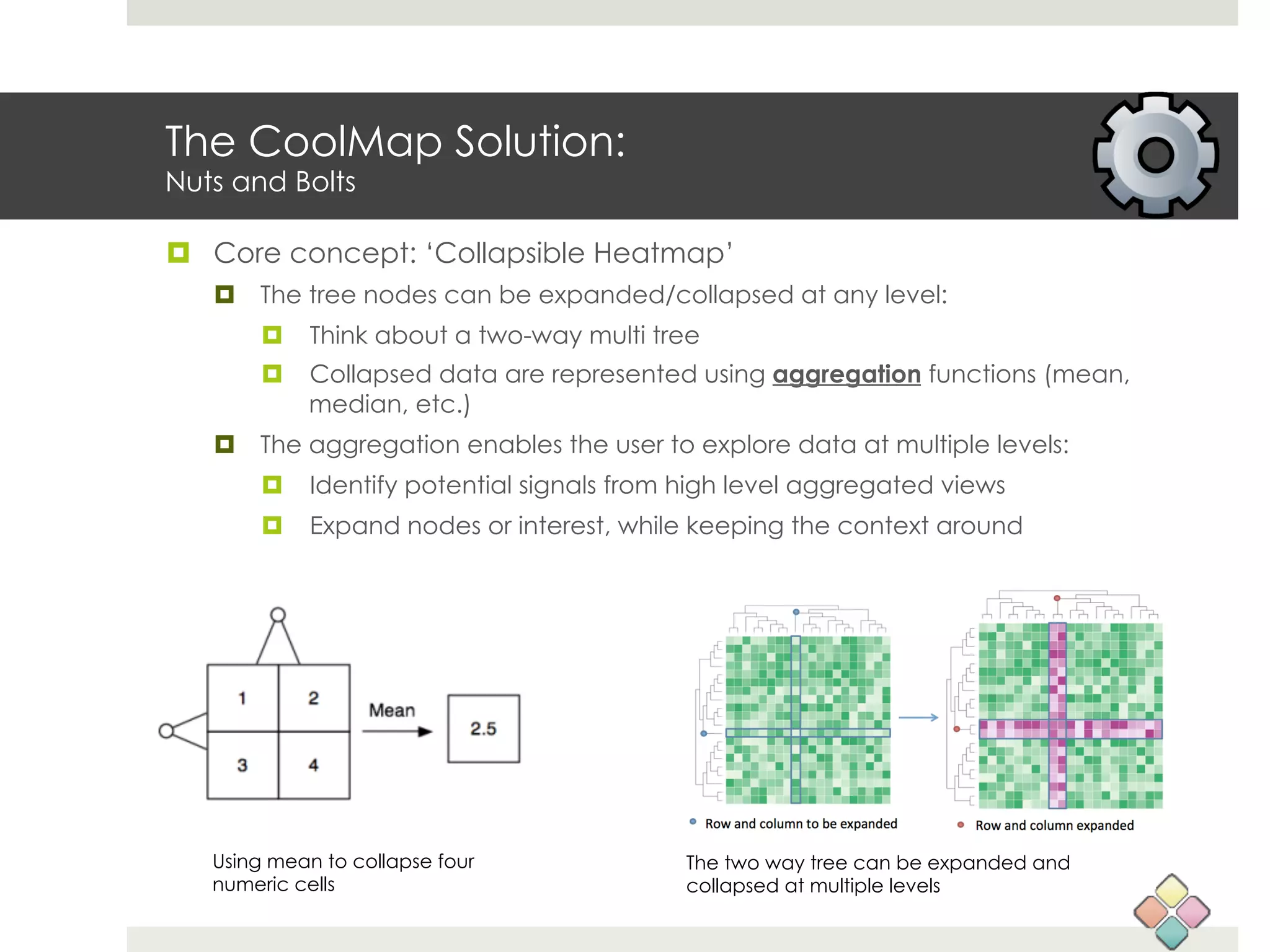
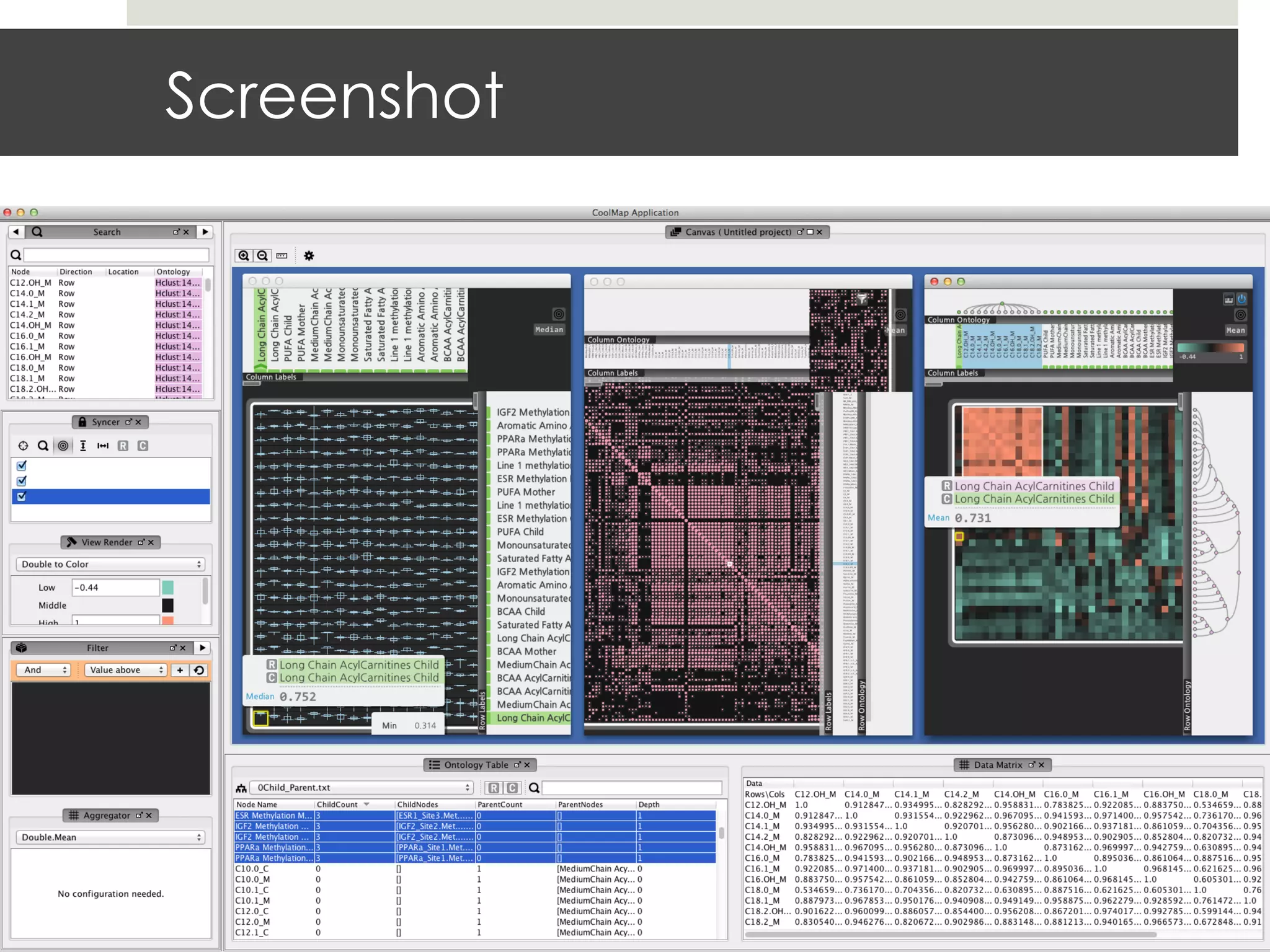
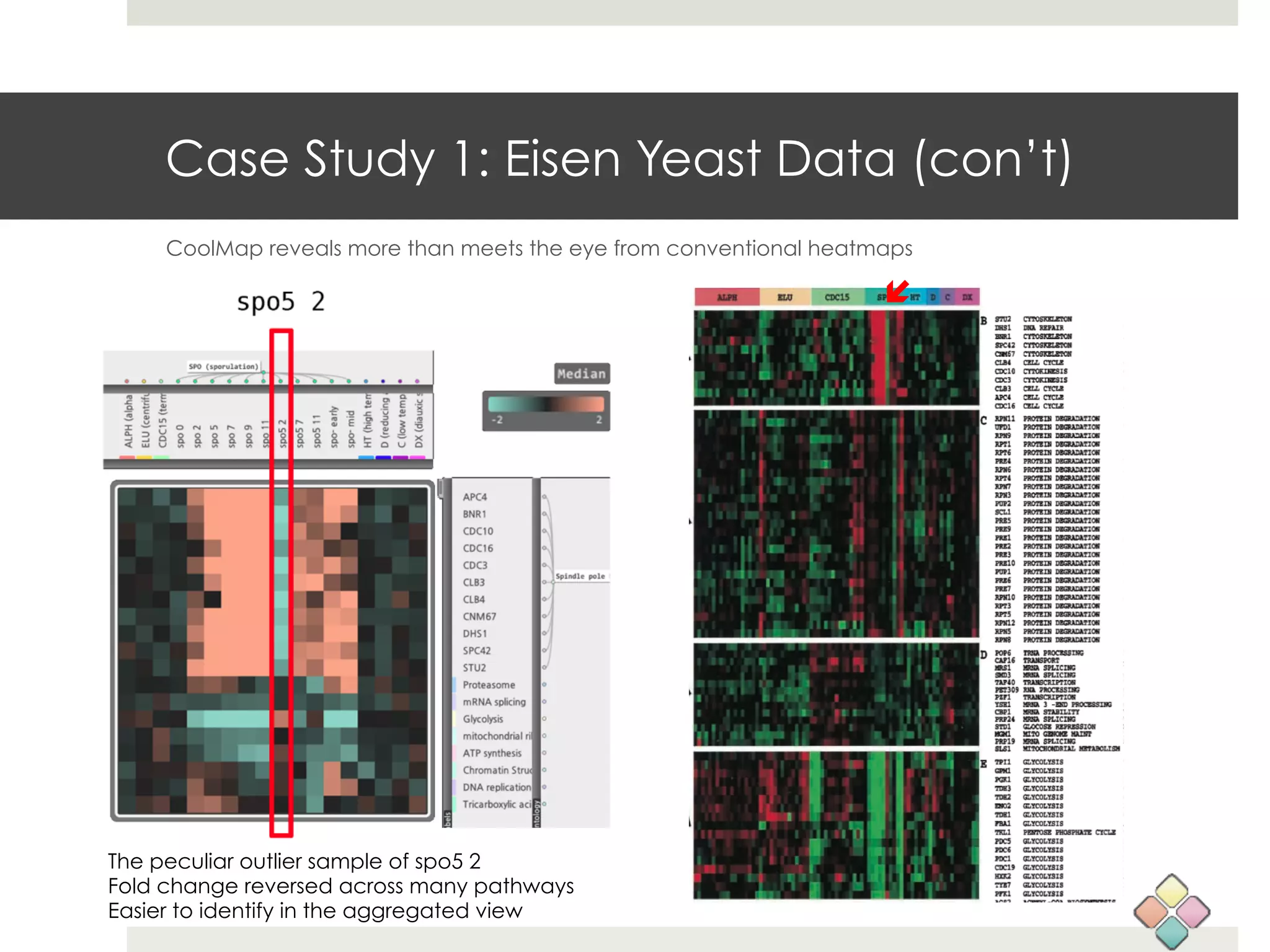
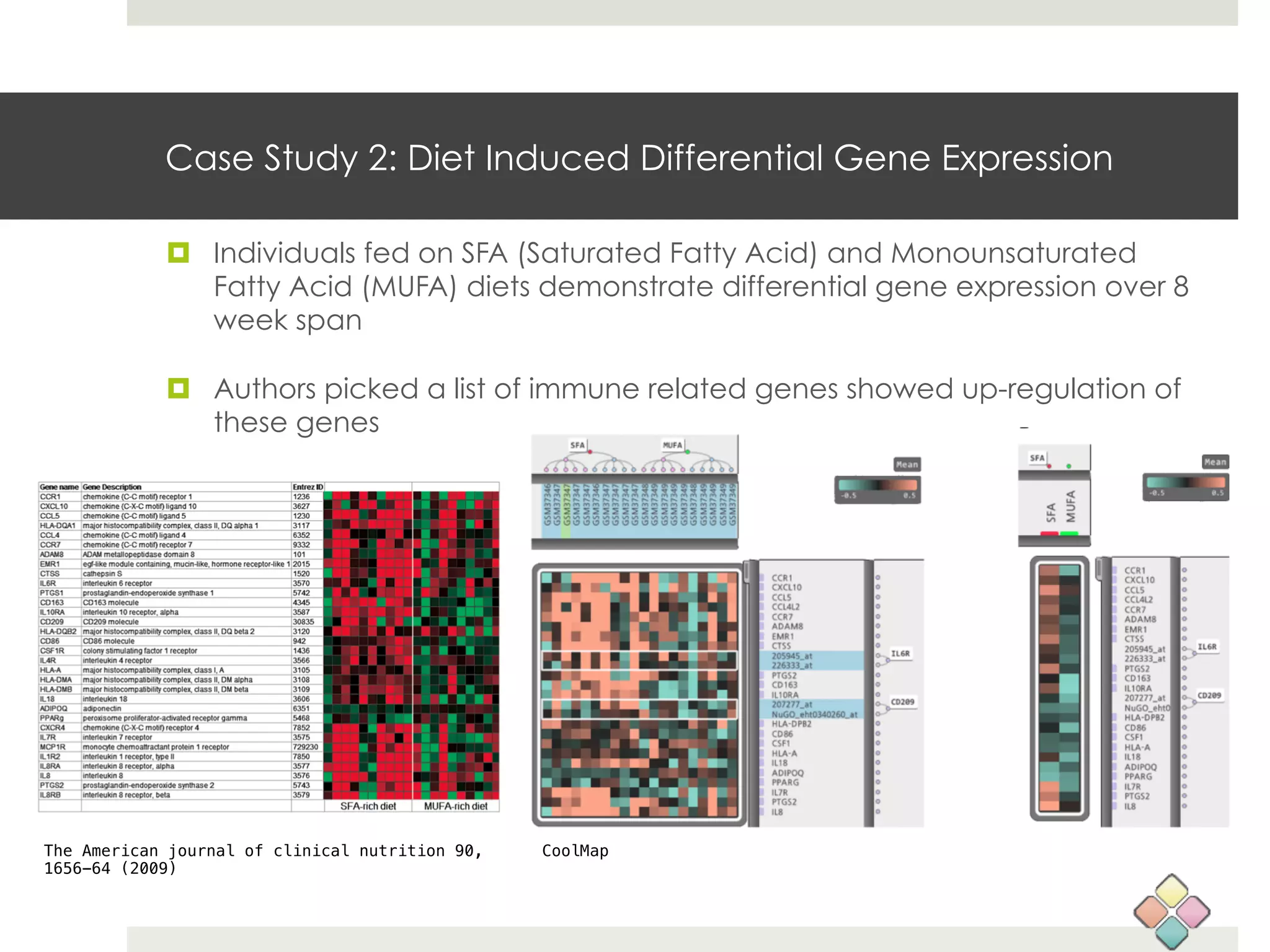
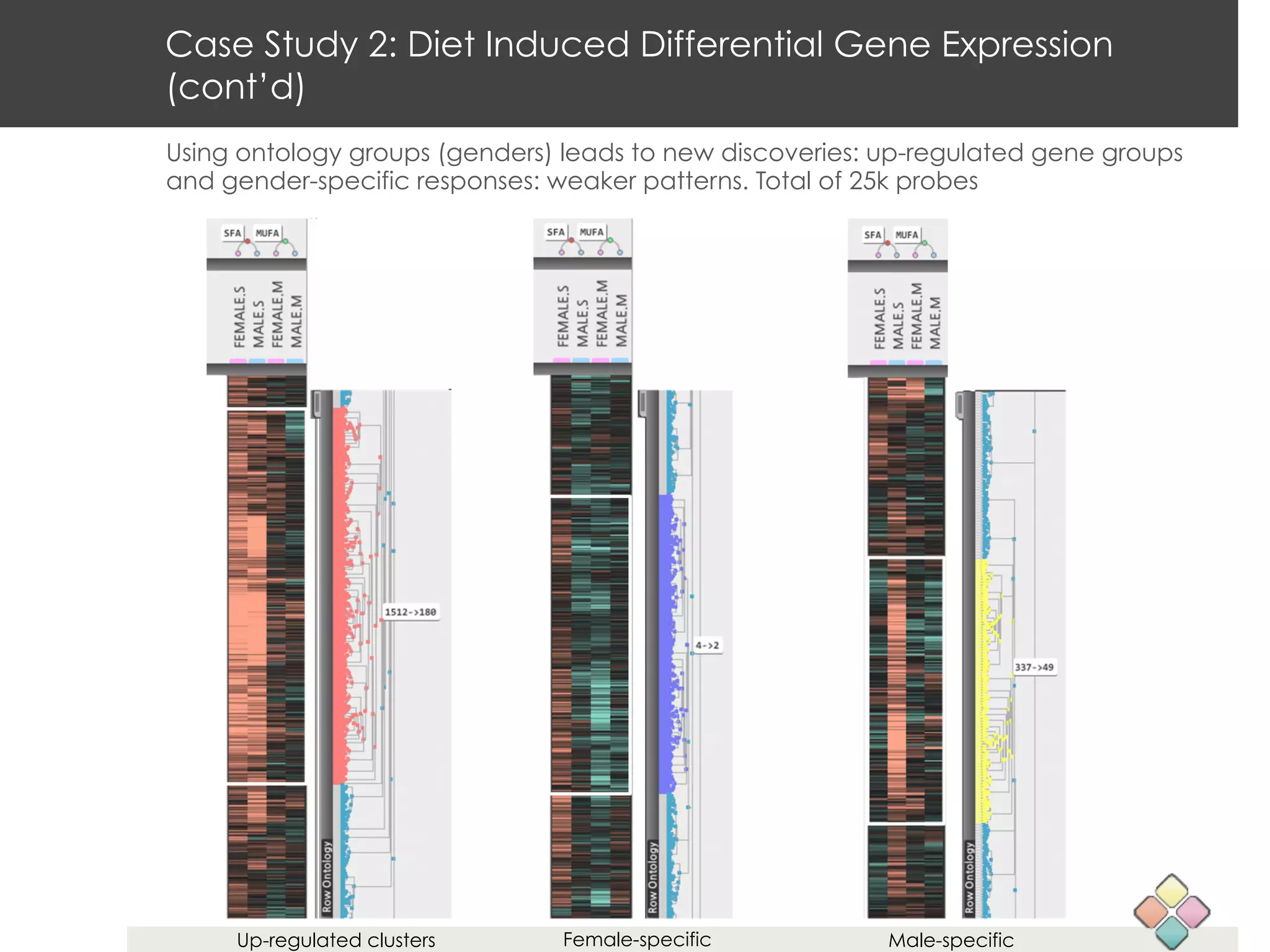
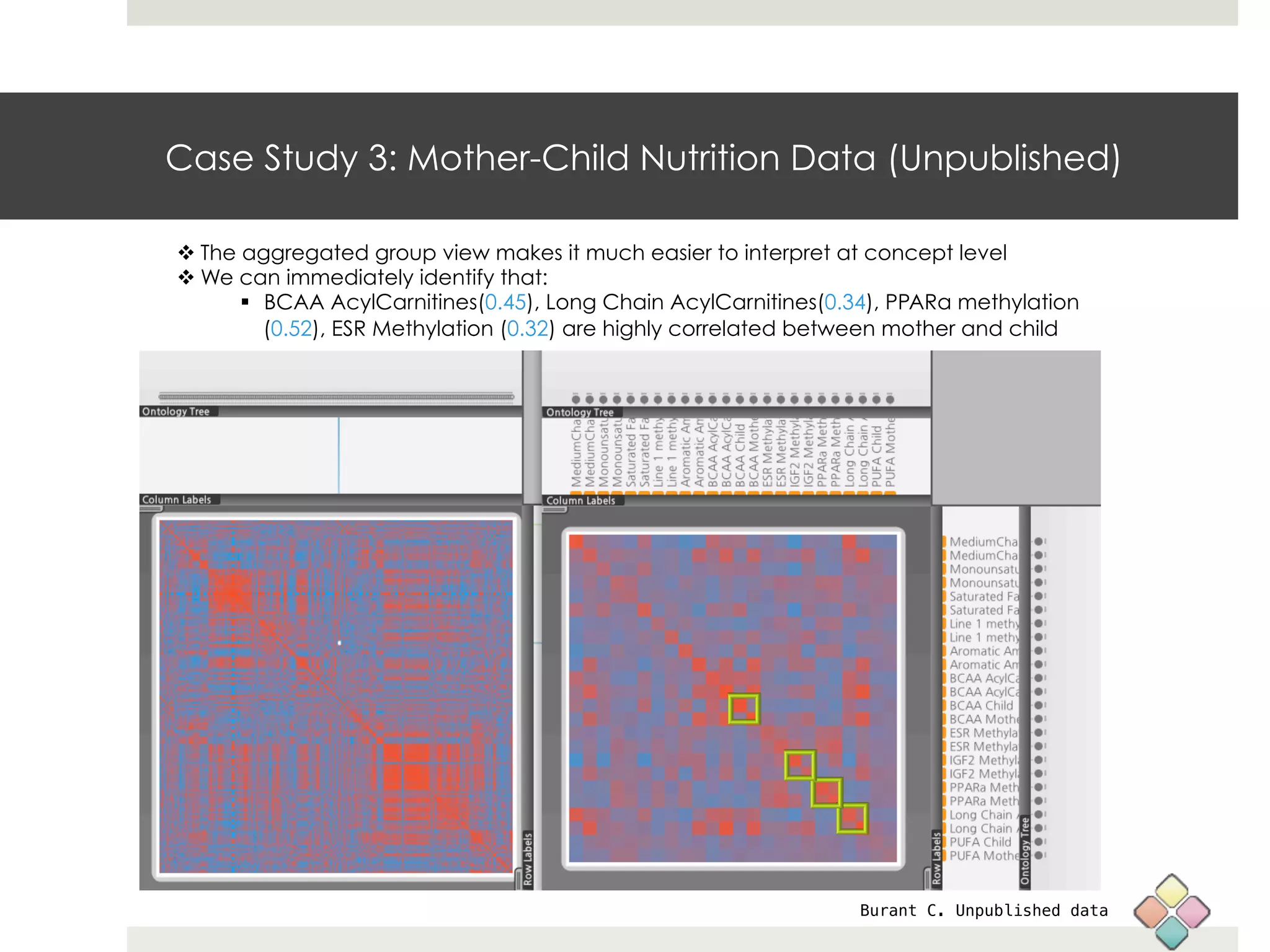
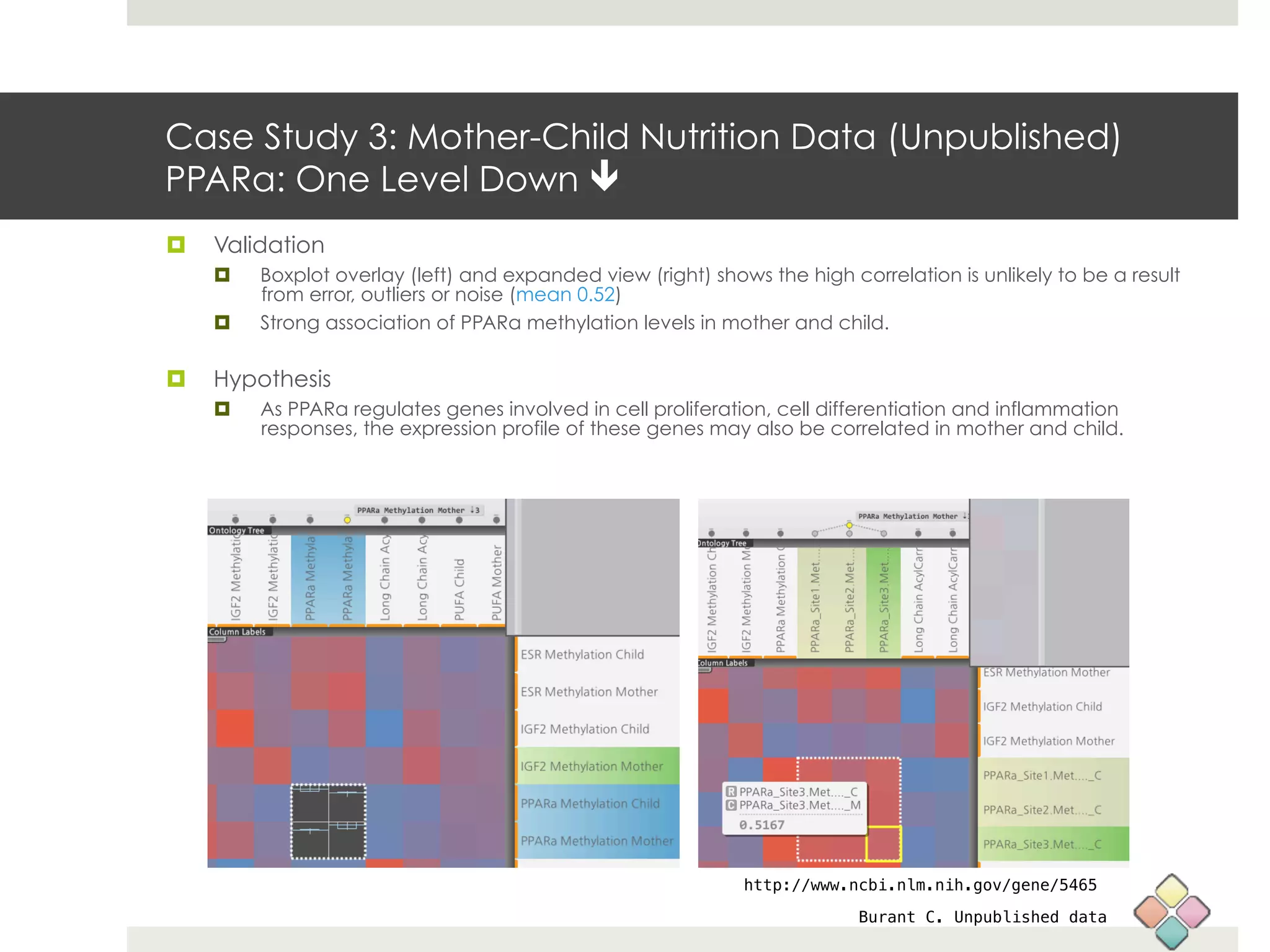
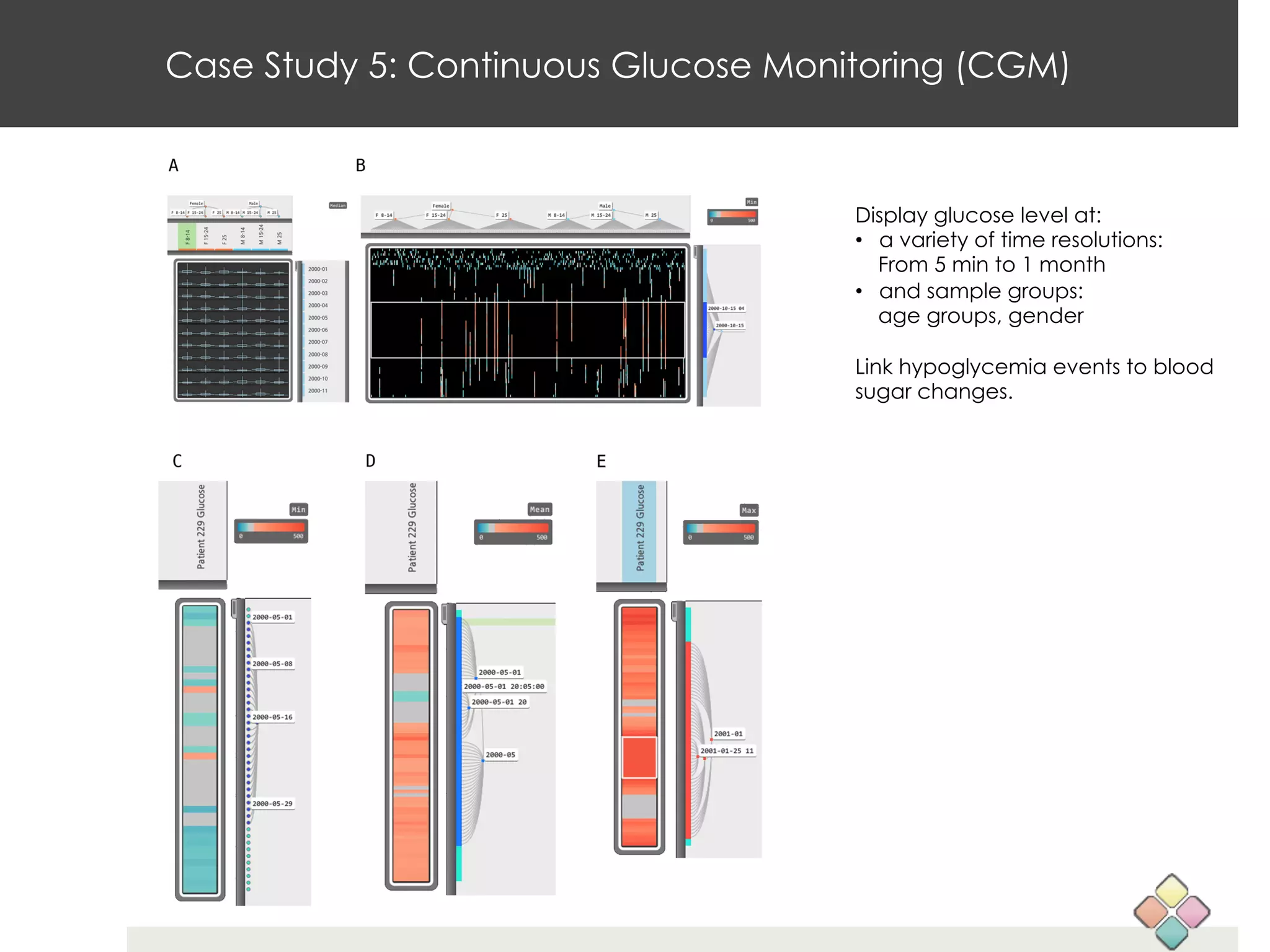
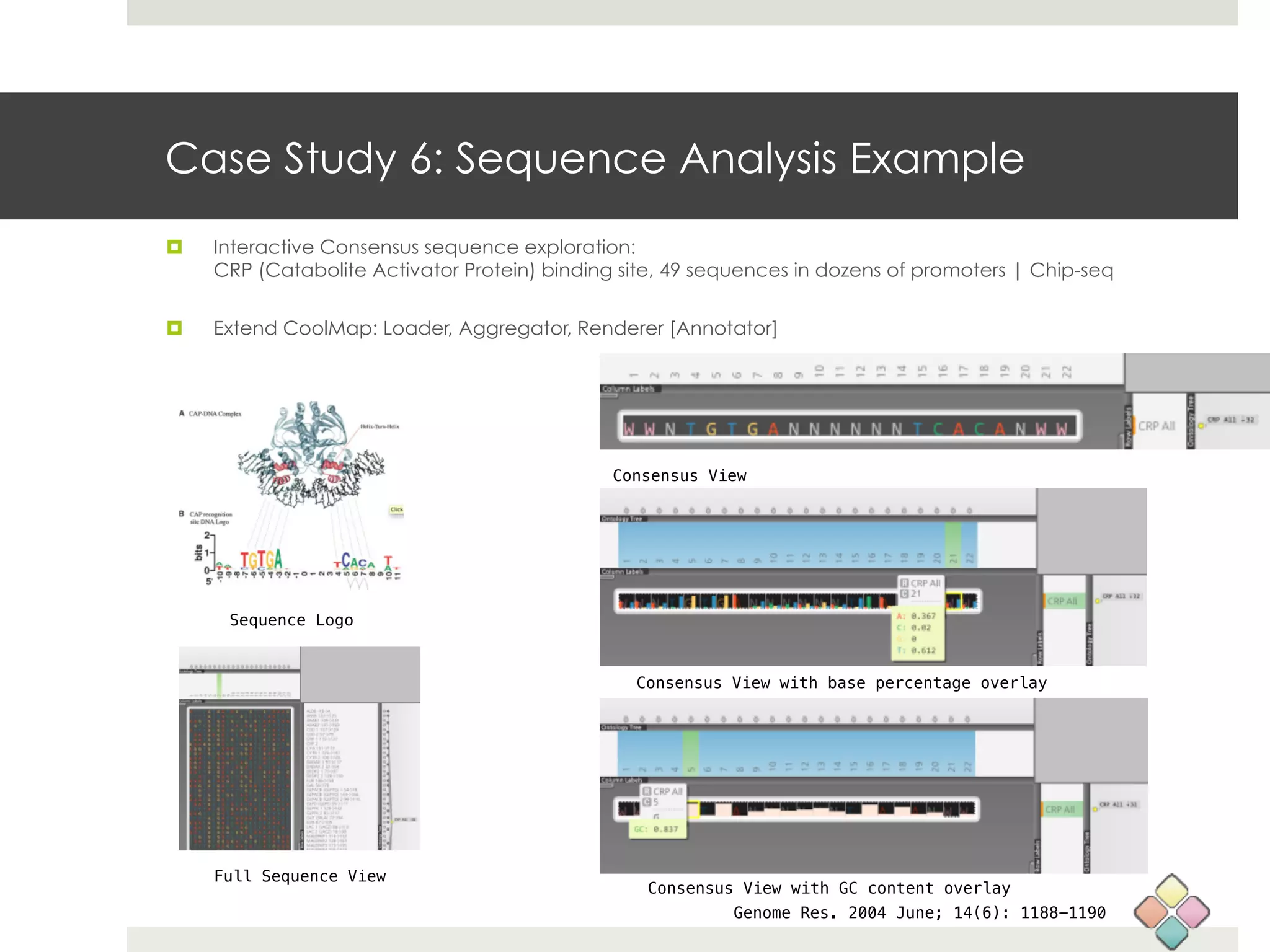
This document describes a new type of heatmap called a "CoolMap" that allows for flexible multi-scale exploration of molecular network data. CoolMaps allow data to be collapsed and aggregated at different levels of a hierarchical tree, enabling visualization and pattern discovery across scales. This approach addresses limitations of conventional heatmaps and enables linking data to existing biological knowledge. Several case studies demonstrate how CoolMaps can provide new insights into gene expression, nutrition, DNA methylation, glucose monitoring, and network data. The core concepts and near-ready software releases are presented, along with acknowledgments.
































![Community Finding with Applications on Phylogenetic Networks [Extended Abstract]](https://cdn.slidesharecdn.com/ss_thumbnails/extendedabstract-190703140727-thumbnail.jpg?width=640&height=640&fit=bounds)




























































