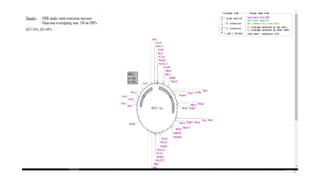
NebCutter is a tool for analyzing DNA sequences to identify restriction enzyme sites and open reading frames (ORFs). It allows users to input sequences from various sources and customize the enzymes for digestion, displaying a schematic of the sequence with relevant information. Users can calculate the sizes of fragments produced by specific enzymes through a simple formula involving base positions.