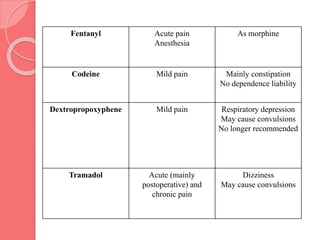
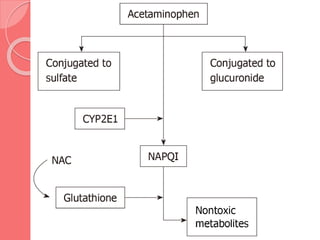
This document summarizes narcotic and non-narcotic analgesics. It describes major classes of analgesics including opioids, NSAIDs, acetaminophen, flupirtine, and ziconotide. Specific drugs are discussed including morphine, methadone, fentanyl, codeine, tramadol, acetaminophen, NSAIDs, flupirtine and ziconotide. Their mechanisms of action, uses, adverse effects, and classification as pure agonists, partial agonists, antagonists are summarized.













![ Morphine rapidly enters all body tissues, including
the fetuses of pregnant women. It should not be used
for analgesia during labor. Infants born to addicted
mothers show physical dependence on opioids and
exhibit withdrawal symptoms if opioids are not
administered.
Morphine is conjugated with glucuronic acid in the
liver to two active metabolites (morphine-6-
glucuronide [M6G] and morphine-3-glucuronide
[M3G]), which are renally excreted.](https://image.slidesharecdn.com/narcoticsandnon-narcoticsanalgesics-220909172601-759272b8/85/Narcotics-and-Non-Narcotics-Analgesics-pptx-14-320.jpg)









































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)




![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

