Download to read offline







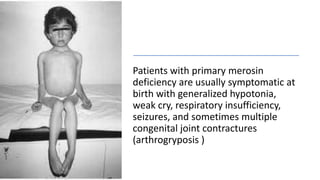
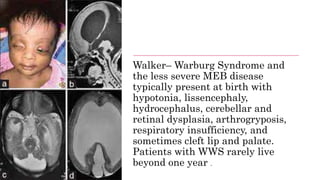
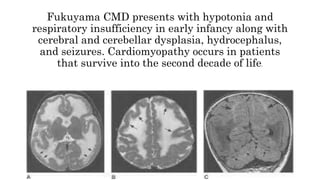




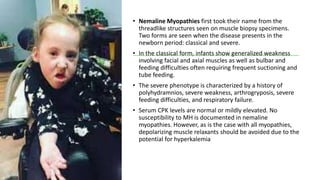
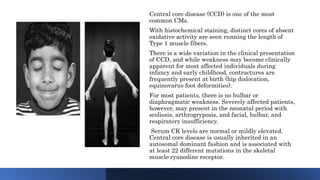

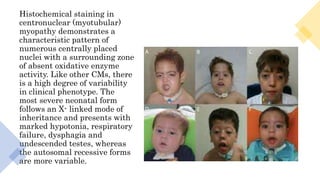
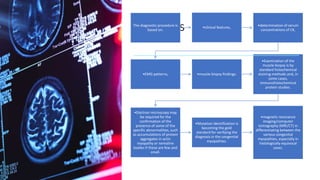





Myopathy is a muscle condition characterized by weakness, which can be congenital or acquired, and includes a range of disorders such as muscular dystrophies and congenital myopathies. These disorders typically present in infancy with symptoms like hypotonia, respiratory difficulties, and developmental delays, with various subtypes identified based on histological features. Diagnosis involves clinical evaluation, CK serum levels, EMG patterns, muscle biopsy, and increasingly, genetic testing.
















![2009 Convegno Malattie Rare Barisoni [23 01]](https://cdn.slidesharecdn.com/ss_thumbnails/2009convegnomalattierarebarisoni2301-090330153020-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





































































