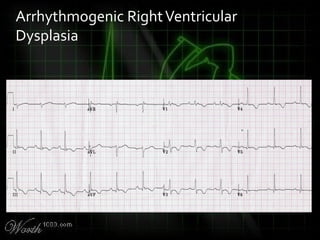
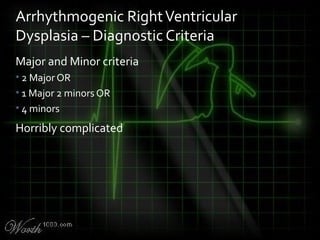
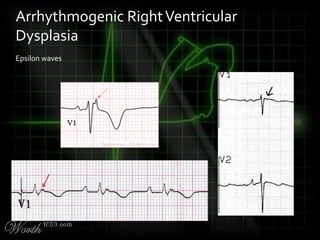
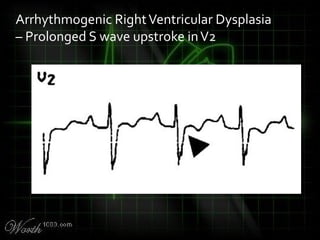
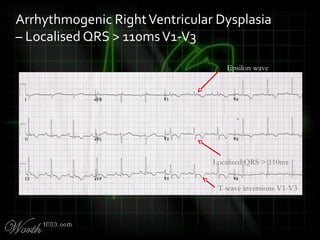
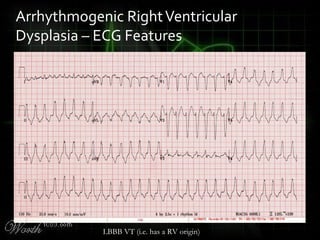
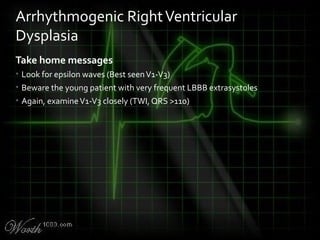
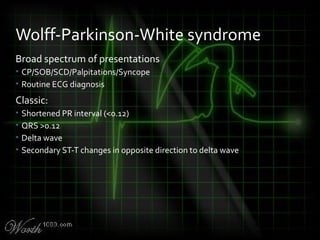
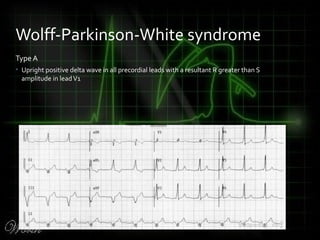
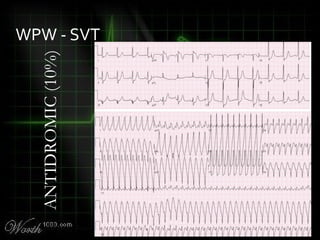
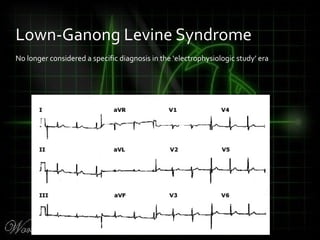
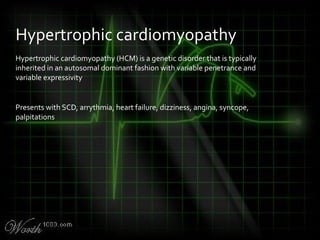
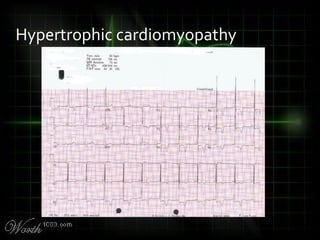
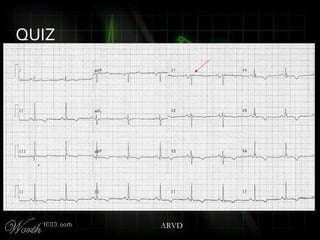
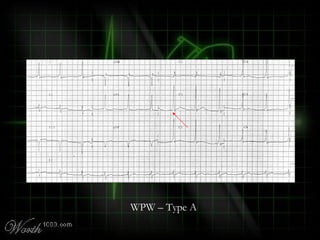
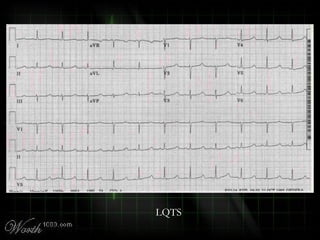
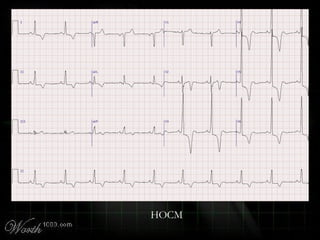
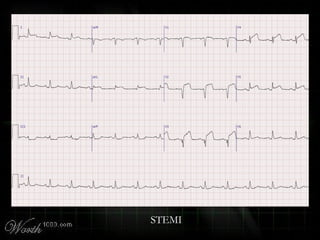
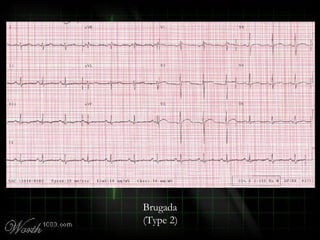
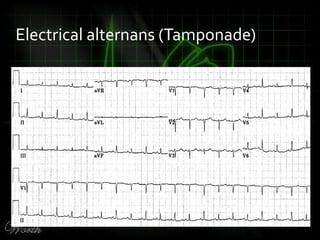
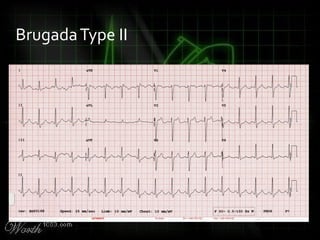
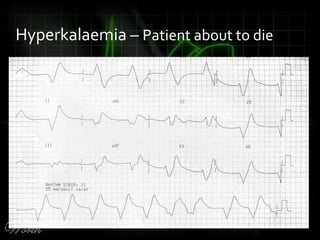
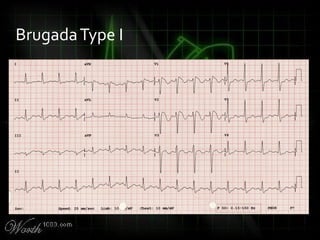
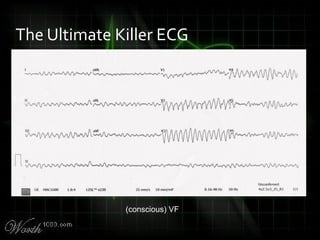
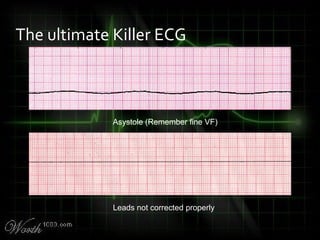
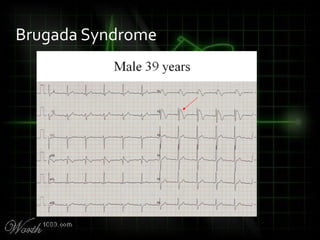
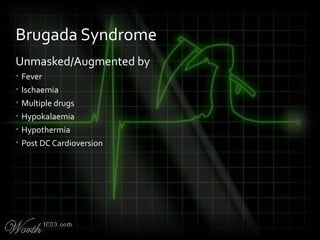
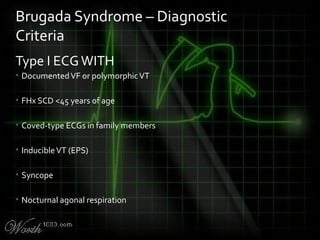
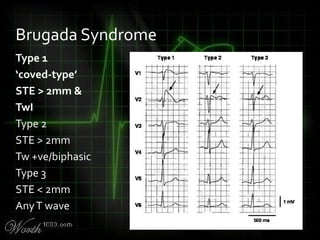
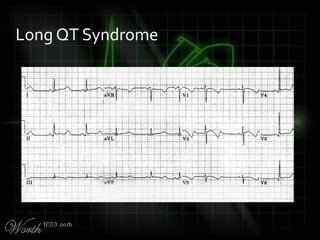
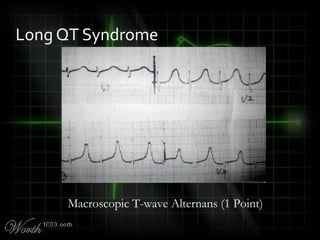
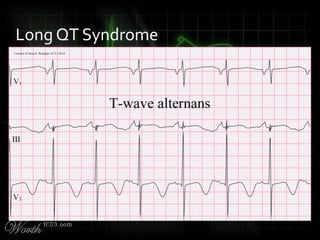
This document provides an overview of various cardiac arrhythmia syndromes including Brugada syndrome, long QT syndrome, short QT syndrome, arrhythmogenic right ventricular dysplasia, Wolff-Parkinson-White syndrome, and hypertrophic cardiomyopathy. It discusses the clinical features, ECG findings, diagnostic criteria, and disposition for each condition. Key advice includes carefully examining leads V1-V3 for abnormalities, being aware of rare conditions like short QT syndrome, and consulting cardiology when these complex syndromes are suspected.












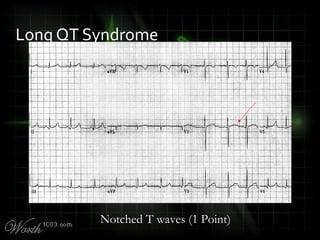
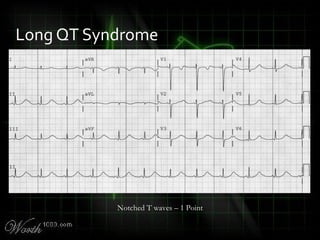


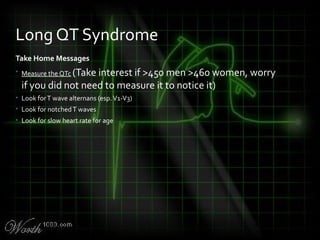
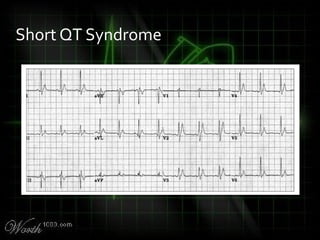



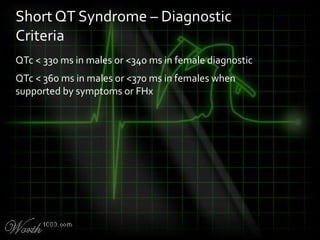
![QT Syndromes – Diagnostic Criteria
Reproduced from Viskin
Viskin S. The QT interval: too long, too short or just right. Heart Rhythm.
2009 May;6(5):711-5. Epub 2009 Mar 3. [PMID: 19389656] [Full text]](https://image.slidesharecdn.com/killerecgs-130917025946-phpapp02/85/Killer-ECGs-33-320.jpg)