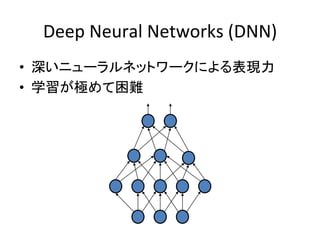
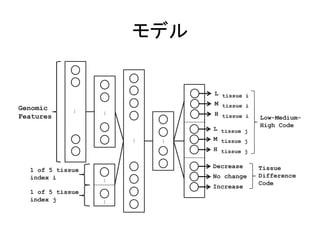
This document summarizes an experiment using deep neural networks (DNNs) to predict alternative splicing patterns in mouse tissues from RNA-seq data. The DNN model contains three hidden layers and jointly represents genomic sequence features and tissue types to predict splicing percentages and changes across tissues. Hyperparameters were optimized using 5-fold cross-validation on AUC. The trained DNN was able to accurately predict splicing patterns for 11,019 exons in 5 mouse tissues, outperforming previous models like Bayesian neural networks and multinomial logistic regression.






![Deep
Neural
Networks
(DNN)
• いくつかのブレークスルー
– Autoencoderによるpre-‐training
[Hinton
et
al.,
2006]
– Dropoutによる学習の安定化 [Srivastava
et
al.,
2014]
• 様々な分野のコンテストで圧倒的な成績
– 画像認識、音声認識、化合物の活性予測、…
• バイオインフォマティクス分野での応用はまだ
それほど多くない
– タンパク質コンタクトマップ予測 [Eickholt
et
al.,
2012]](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-8-320.jpg)
![DNNのプレトレーニング
Stacked
Autoencoder
‡ 層ごとにオートエンコーダを学習→ 過学習を克服
± “greedy layerwise pretraining” [Hinton06]
スパースオートエンコーダ
‡ 入力サンプルをよく再現するように
± BPでor ボルツマンマシンとして学習
± 中間層がスパースに活性化するように正則化を行う
[岡谷,
2013]
• 層ごとに教師なし学習
• 各層は入力をよく再現するように学習](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-9-320.jpg)
![Dropout
• ランダムに隠れユニットを取り除いて学習
Srivastava, Hinton, Krizhevsky, Sutskever and Salakhutdinov
• アンサンブル学習と同じ効果
(a) Standard Neural Net (b) After applying dropout.
Figure 1: Dropout Neural Net Model. Left: A standard neural net with 2 hidden layers. Right:
[Srivastava
et
al.,
2014]
An example of a thinned net produced by applying dropout to the network on the left.
Crossed units have been dropped.](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-10-320.jpg)
![Deep
Neural
Networks
(DNN)
• 深いニューラルネットワークによる表現力
Neural Networks
hidden layers
not
initialized)
perform
Different Levels of Abstraction
• Hierarchical Learning
– Natural progression from low
level to high level structure as
seen in natural complexity
– Easier to monitor what is being
learnt and to guide the machine
to better subspaces
– A good lower level
representation can be used for
many distinct tasks
[Lee,
2010]](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-11-320.jpg)
![問題設定
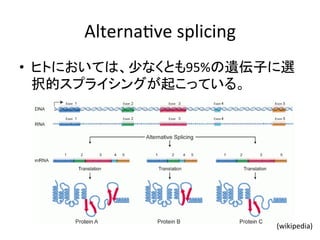
ARTICLES • エクソンがスプライシングを受けるかどうかを
予測する
features, active Information We use theory31 about the code. better than improved To assemble the compendium, parameters 300 nt 300 nt 300 nt 300 nt
RNA feature
extraction
Splicing code
5). The but diminished (Fig. 1b, code contained,a
Tissue type
Alternatively spliced exon
Feature set
Predicted change in
exon inclusion Code assembly
b
[Barash
et
al.,
2010]](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-12-320.jpg)
![特徴量
• 前後のエクソン・イントロンに関する1392個の特
徴量[Barash
et
al.,
2010]
300 nt 300 nt 300 nt 300 nt
ARTICLES – k-‐mer
– 翻訳可能性
– 長さ
– 保存度
– モチーフ配列(転写因子結合部位)
– …
features, thresholds active feature Information We use a measure theory31 (see Methods). about genome-the code. A code better than guessing, improved prediction To assemble a the compendium, parameters to maximize 5). The code but diminished gains (Fig. 1b, c, based code Splicing code
contained,compendium plus did not exceed 1 a
Tissue type
Alternatively spliced exon
Feature set
Predicted change in
exon inclusion bits)
400
Final
assembled
(code
d
b
RNA feature
extraction
Code assembly](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-14-320.jpg)
![出力
• PSI
(Percentage
of
Splicing
In)
[Katz
et
al.,
2010]を離
散化
– LMH
code
• Low:
0-‐0.33,
Medium:
0.33-‐0.66,
High:
0.66-‐1
– DNI
code
• Decrease:
部位i
>
部位j
• No
change:
部位i
≒
部位j
(PSIの差の絶対値<0.15)
• Increase:
部位i
<
部位j
• 複数の出力を同時に学習
– 学習が安定化
1. Deep learning Architecture of the DNN used to predict AS patterns. It contains three hidden layers, with hidden variables that jointly](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-15-320.jpg)

![perform well on both tasks. The optimal set of hyperparameters were then used model using both training and validation data. Five models were trained this different folds of data. Predictions made for the corresponding test data from all then evaluated and reported.
The hyperparameters that were optimized and their search ranges are: (1) the learning each of the two tasks (0.1 to 2.0), (2) the number of hidden units in each layer (30 the L1 penalty (0.0 to 0.25), (4) the standard deviation of the normal distribution initialize the weights (0.001 to 0.200), (5) the momentum schedule defined as epochs to linearly increase the momentum from 0.50 to 0.99 (50 to 1500), and (6) size (500 to 8500). The number of training epoch was fixed to 1500. In our experience, set of hyperparameters were generally found in approximately 2 days, where experiments ran on a single GPU (Nvidia GTX Titan). The selected set of hyperparameters Table S2. There is a large range of acceptable values for the number hidden units layer.
ハイパーパラメータの最適化
• 5-‐fold
cross
validaDon:
AUCに基づき最適化
– training:
3
folds
(DNNの学習)
– validaDon:
1
fold
(ハイパーパラメータの最適化)
– test:
1
fold
(評価)
Table S2. The hyperparameters selected to train the deep neural network. Some ranges to reflect the variations from the different folds as well as hyperparameters performing runs within a given fold.
• ガウス過程に基づく
spearmint
[Snoek
et
al.,
2012]
という手法を適用
Range Selected
Hidden Units (layer 1) 450 - 650
Hidden Units (layer 2) 4500 - 6000
Hidden Units (layer 3) 400 - 600
L1 Regularization 0 - 0.05
Learning Rate (LMH code) 1.40 - 1.50
Learning Rate (DNI code) 1.80 - 2.00
Momentum Rate 1250
Minibatch Size 1500
Weight Initialization 0.05 - 0.09](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-17-320.jpg)
![Toronto, ON, Canada M5S 3G4
実験
• 実験環境
– Python
with
Gnumpy
[Tieleman,
2010]
で実装
– Nvidia
GTX
Titan上で実験
• データ
– マウスの5部位RNA-‐seqデータ [Brawand
et
al.,
2011]
か
ら得た
11,019個のエクソンのスプライシングパ
ターン
1
S1 Dataset Description
The dataset consists of 11,019 mouse alternative exons in five tissue types profiled from RNA-Seq
data prepared by (Brawand et al., 2011). As explained in the main text, a distribution of
percent-spliced-in (PSI) was estimated for each exon and tissue. From this distribution, three
real-values were calculated by summing the probability mass over equally split intervals of 0 to
0.33 (low), 0.33 to 0.66 (medium), and 0.66 to 1 (high). They represent the probability that the
given exon within a tissue type has PSI value ranging from these intervals, hence are soft
assignments into each category. The models were trained using these soft labels. Table S1
shows the distribution of exons in each category, counted by selecting the label with the largest
value.
Table S1. The number of exons classified as low, medium, and high for each mouse tissue.
Exons with large tissue variability (TV) are displayed in a separate column. The proportion of
medium category exons that have large tissue variability is higher than the other two categories.
Brain Heart Kidney Liver Testis
All TV All TV All TV All TV All TV
Low 1782 579 1191 460 1287 528 1001 413 1216 452
Medium 669 456 384 330 345 294 254 220 346 270
High 5229 1068 4060 919 4357 941 3606 757 4161 887
Total 7680 2103 5635 1709 5989 1763 4861 1390 5723 1609](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-18-320.jpg)
![Heart MLR 84.6"0.1 73.1"0.3 83.6"0.1
Downloaded from http://bioinformatics.oxfordjournals.3 We present three sets of results that compare the test perform-ance
BNN 89.2"0.4 75.2"0.3 88.0"0.4
DNN 89.3"0.5 79.4"0.9 88.3"0.6
BNN 91.1"0.3 74.7"0.3 89.5"0.2
DNN 90.7"0.6 79.7"1.2 89.4"1.1
結果:先行研究との比較
RESULTS
of the BNN, DNN and MLR for splicing pattern predic-tion.
The first is the PSI prediction from the LMH code tested on
all exons. The second is the PSI prediction evaluated only on
targets where there are large Deep variations learning across of the tissues splicing for a code
given
exon. These are events where "PSI!"0.15 for at least one pair
of tissues, third result • LMH
to evaluate the tissue specificity of the model. The
shows how code
well the code (all)
can classify "PSI between
the five tissue types. Hyperparameter tuning was used in all
methods. The averaged predictions from all partitions and
folds are used to evaluate the model’s performance on their cor-responding
Kidney MLR 86.7"0.1 75.6"0.2 86.3"0.1
BNN 92.5"0.4 78.3"0.4 91.6"0.4
DNN 91.9"0.6 82.6"1.1 91.2"0.9
Liver MLR 86.5"0.2 75.6"0.2 86.5"0.1
BNN 92.7"0.3 77.9"0.6 92.3"0.5
DNN 92.2"0.5 80.5"1.0 91.1"0.8
• LMH
code
(high
Dssue
Testis MLR 85.6"0.1 72.3"0.4 85.2"0.1
BNN 91.1"0.3 75.5"0.6 90.4"0.3
DNN 90.7"0.6 76.6"0.7 89.7"0.7
variability)
Table 1. Comparison of the LMH code’s AUC performance on different
methods
(a) AUCLMH_All
test dataset. Similar to training, we tested on exons
and tissues that have at least 10 junction reads.
For the LMH code, as the same prediction target can be gen-erated
Tissue Method Low Medium High
by different input configurations, and there are two LMH
Brain MLR 81.3"0.1 72.4"0.3 81.5"0.1
outputs, we BNN compute the 89.2predictions "0.4 for 75.2all "input 0.3 combinations
88.0"0.4
containing DNN the particular 89.3tissue "0.5 and average 79.4"them 0.9 into 88.3a single
"0.6
prediction for testing. To assess the stability of the LMH predic-tions,
Heart MLR 84.6"0.1 73.1"0.3 83.6"0.1
BNN 91.1"0.3 74.7"0.3 89.5"0.2
DNN 90.7"0.6 79.7"1.2 89.4"1.1
we calculated the percentage of instances in which there is
a prediction from one tissue input configuration that does not
agree with another tissue input configuration in terms of class
membership, for all exons and tissues. Of all predictions, 91.0%
agreed with each other, 4.2% have predictions that are in adja-cent
Kidney MLR 86.7"0.1 75.6"0.2 86.3"0.1
BNN 92.5"0.4 78.3"0.4 91.6"0.4
DNN 91.9"0.6 82.6"1.1 91.2"0.9
Liver MLR 86.5"0.2 75.6"0.2 86.5"0.1
classes (i.e. low and medium, or medium and high), and 4.8%
BNN 92.7"0.3 77.9"0.6 92.3"0.5
DNN 92.2"0.5 80.5"1.0 91.1"0.8
otherwise. Of those predictions that agreed with each other,
85.9% correspond to the correct class label on test data,
51.2% for the predictions with adjacent classes and 53.8% for
the remaining predictions. This information can be used to assess
the confidence of the predicted class labels. Note that predictions
spanning adjacent classes may be indicative that the PSI value is
somewhere between the two classes, and the above analysis using
hard class labels can underestimate the confidence of the model.
Testis MLR 85.6"0.1 72.3"0.4 85.2"0.1
BNN 91.1"0.3 75.5"0.6 90.4"0.3
DNN 90.7"0.6 76.6"0.7 89.7"0.7
(b) AUCLMH_TV
BNN:Bayeisian
NN
[Xiong
et
al.,
2011],
MLR:
MulDnomial
LogisDc
Regression
Tissue Method Low Medium High
(b) AUCLMH_TV
Tissue Method Low Medium High
Brain MLR 71.1"0.2 58.8"0.2 70.8"0.1
BNN 77.9"0.5 61.1"0.5 76.5"0.7
DNN 82.8"1.0 69.5"1.1 81.1"0.4
Heart MLR 73.9"0.3 58.6"0.4 72.7"0.1
BNN 78.1"0.3 58.9"0.3 75.7"0.3
DNN 82.0"1.1 67.4"1.3 79.7"1.2
Kidney MLR 79.7"0.3 64.3"0.2 79.4"0.2
BNN 83.9"0.5 66.4"0.5 83.3"0.6
DNN 86.2"0.6 73.2"1.3 85.3"1.2
Liver MLR 80.1"0.5 63.7"0.3 79.4"0.3
BNN 84.9"0.7 65.4"0.7 84.4"0.7
DNN 87.7"0.6 69.4"1.2 84.8"0.8
Testis MLR 77.3"0.2 60.8"0.3 77.0"0.1
BNN 81.1"0.5 63.9"0.9 81.0"0.5
DNN 84.6"1.1 67.8"0.9 83.5"0.9
Notes: " indicates 1 standard deviation; top performances are shown in bold.
subset of events that exhibit large tissue variability. Here, the
DNN significantly outperforms the BNN in all categories and](https://image.slidesharecdn.com/ismb2014-140910224126-phpapp02/85/ISMB2014-Deep-learning-of-the-tissue-regulated-splicing-code-19-320.jpg)























![Data driven model optimization [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/datadrivenmodeloptimizationautosaved-180427045633-thumbnail.jpg?width=640&height=640&fit=bounds)
























































