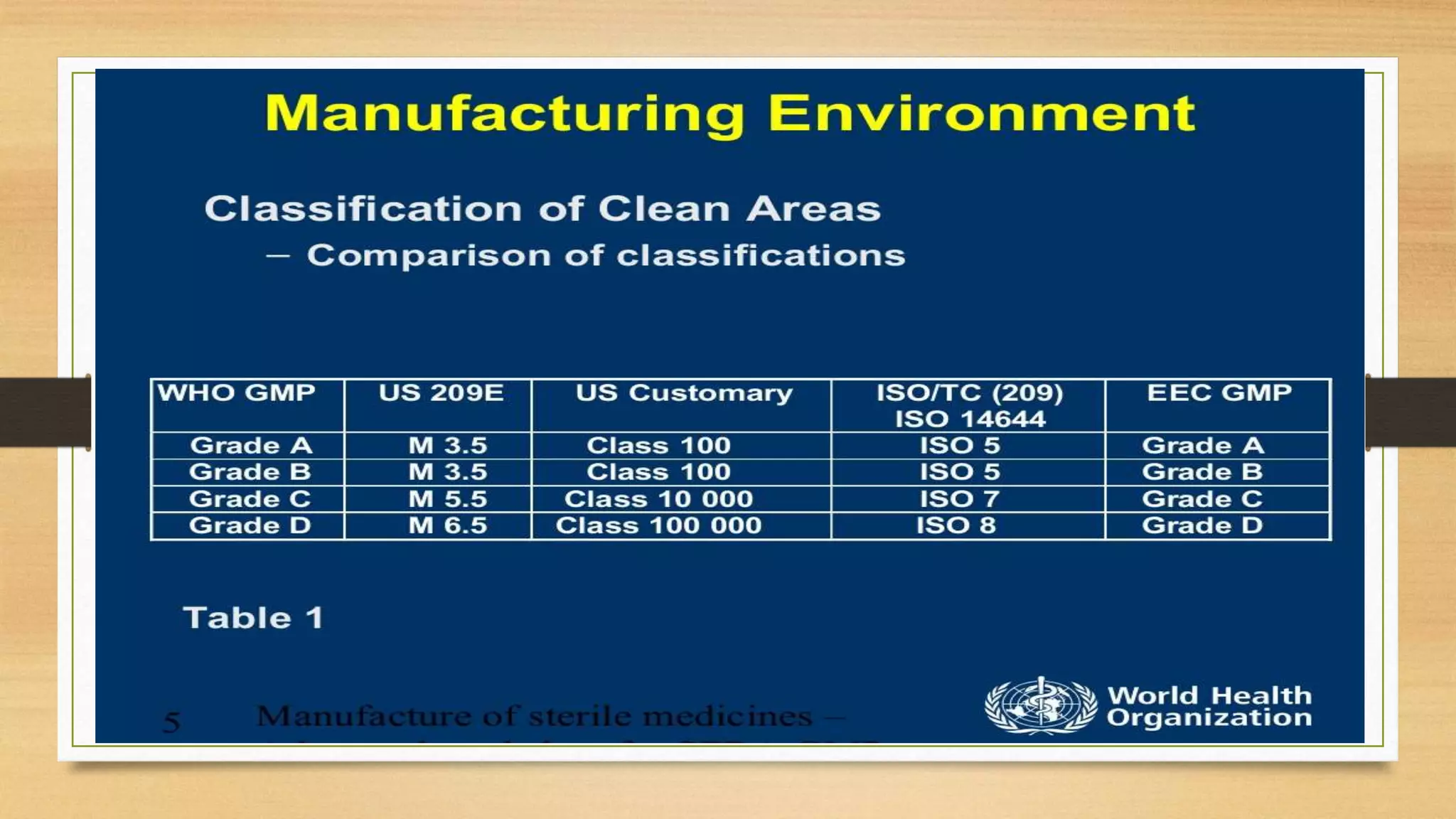
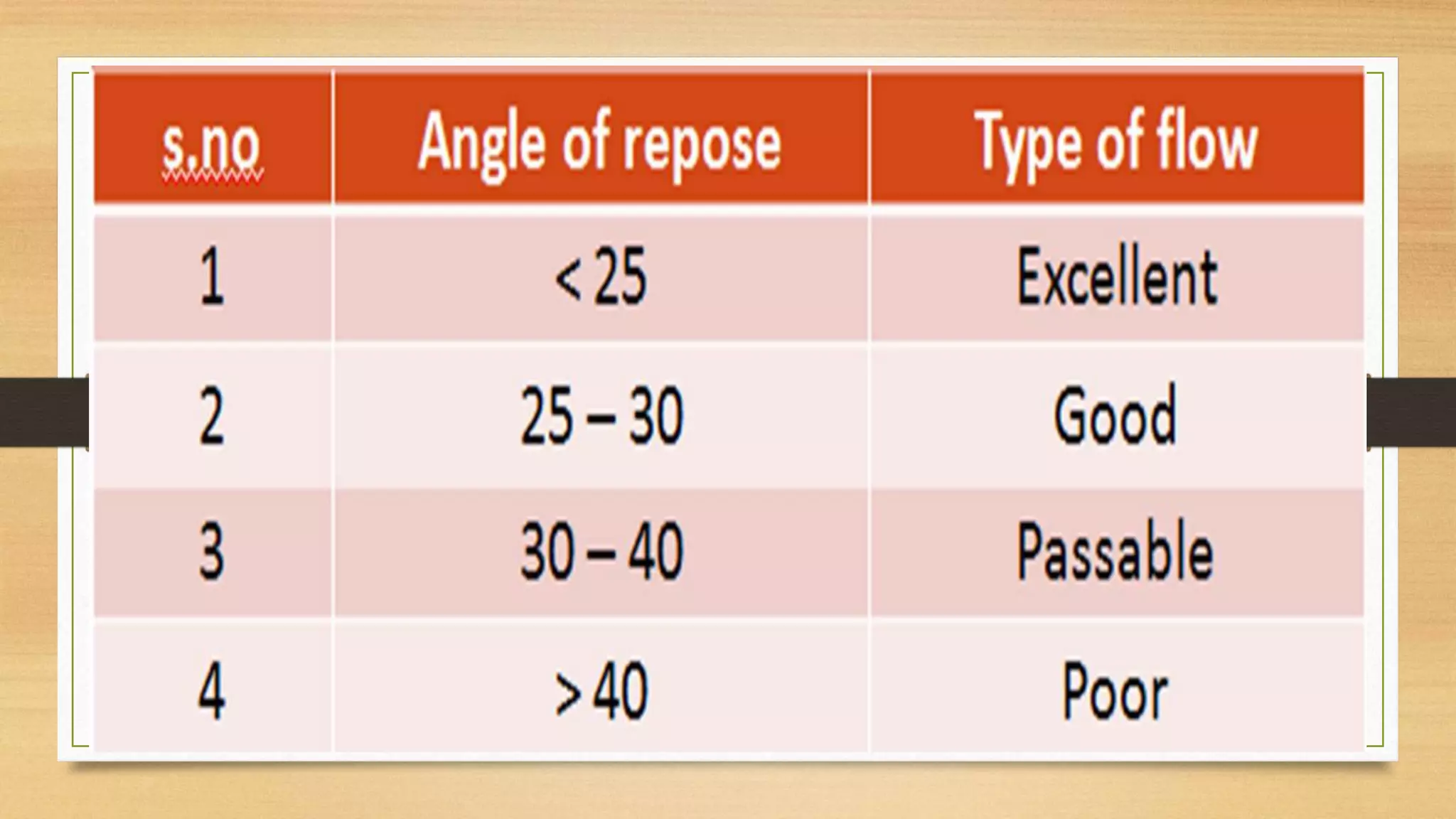
This document provides information on aseptic processing and in-process quality control tests for various dosage forms including ointments, suspensions, emulsions, powders, and parenterals. It describes how sterile products are manufactured through aseptic processing to ensure sterility. It also outlines various quality tests done during manufacturing to monitor product quality, such as appearance, viscosity, particle size, moisture content, clarity, pH, and microbial limits.