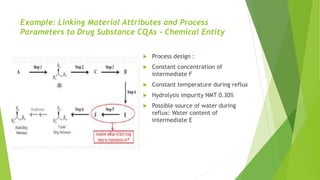
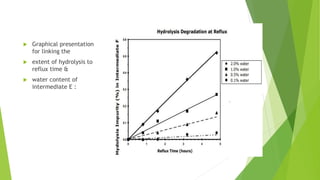
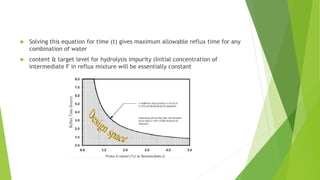
The document outlines the guidelines for the development and manufacture of drug substances in alignment with ICH standards, focusing on ensuring product quality and consistency through a structured control strategy. It discusses the importance of critical quality attributes (CQAs), manufacturing process development, risk management, and the necessity of validating processes to ensure consistent quality in drug substance production. Additionally, it emphasizes the need for ongoing knowledge management throughout the product lifecycle to adapt to changes and maintain quality.













































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)




