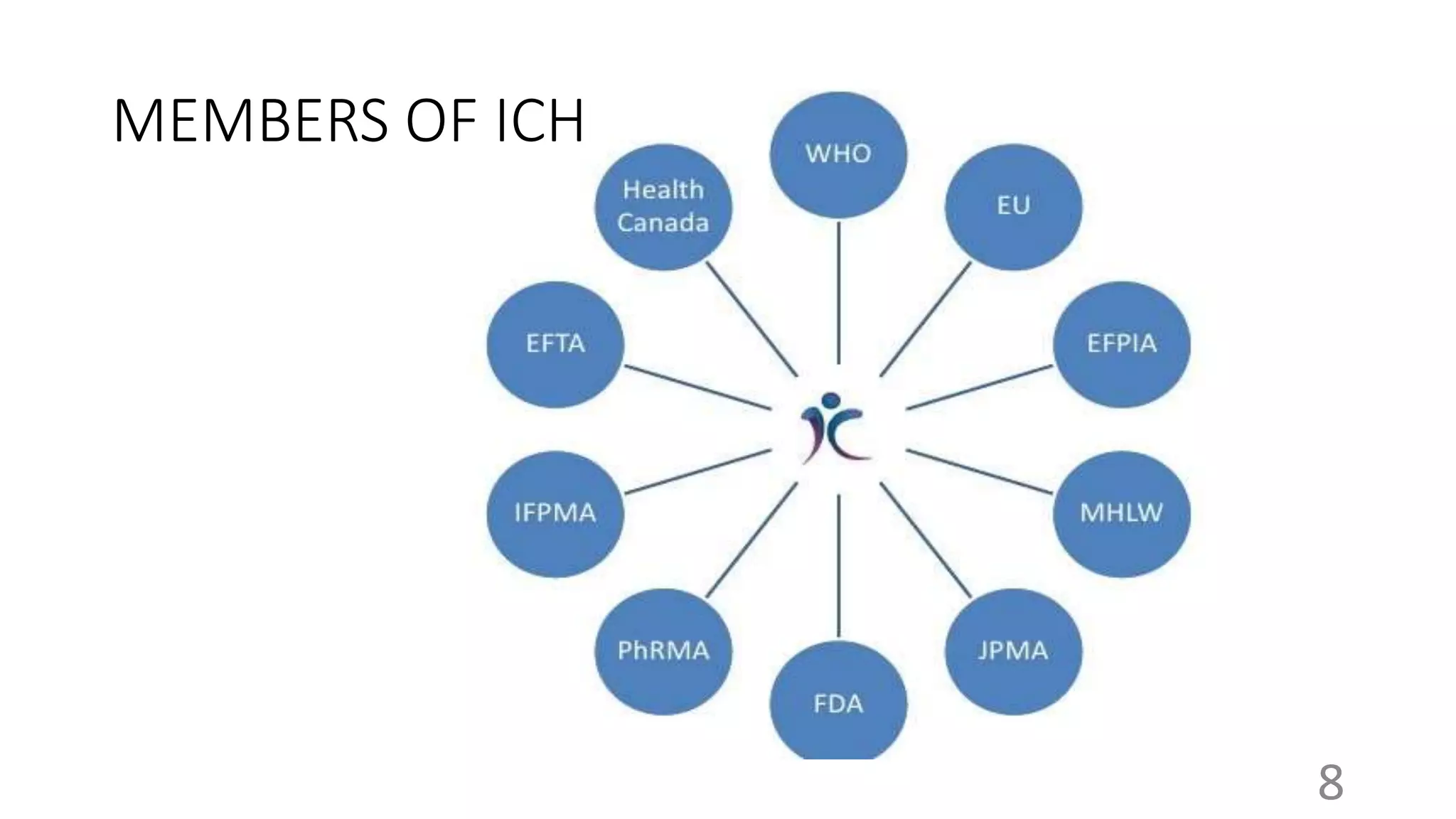
The document provides an overview of the International Council for Harmonisation (ICH), which brings together regulatory authorities and the pharmaceutical industry to discuss scientific and technical aspects of pharmaceuticals and develop guidelines. It discusses the objectives, history, members, organizational structure, and guidelines of ICH. The guidelines cover quality, safety, efficacy, and multidisciplinary topics related to developing, registering, and maintaining safe and effective medicines. Key guidelines include those on stability testing, carcinogenicity studies, clinical trials, and the Common Technical Document format.
















































![cmc [ chemistry manufacturing control ]](https://cdn.slidesharecdn.com/ss_thumbnails/presentation2222ra-181120122336-thumbnail.jpg?width=640&height=640&fit=bounds)


























































