















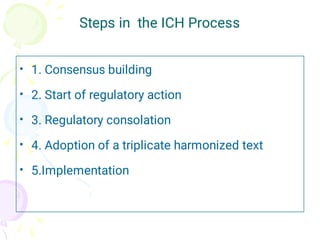










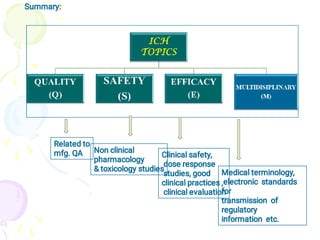




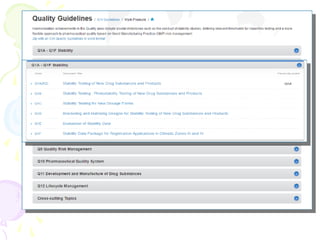
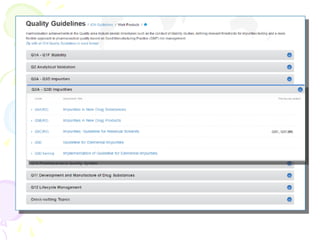



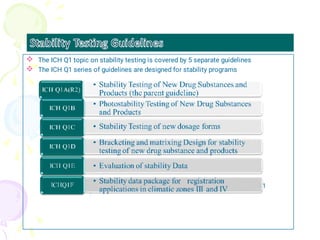















































































The International Council for Harmonization (ICH) brings together regulatory authorities and the pharmaceutical industry from Europe, Japan, and the US to discuss scientific and technical aspects of drug registration. ICH aims to harmonize technical requirements for registration to reduce duplication of testing and help ensure safe, effective, and high quality medicines are developed efficiently. ICH has published guidelines on quality, safety, efficacy, and multidisciplinary topics related to manufacturing, nonclinical studies, clinical trials, and electronic standards.














![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)

































































