Downloaded 16 times

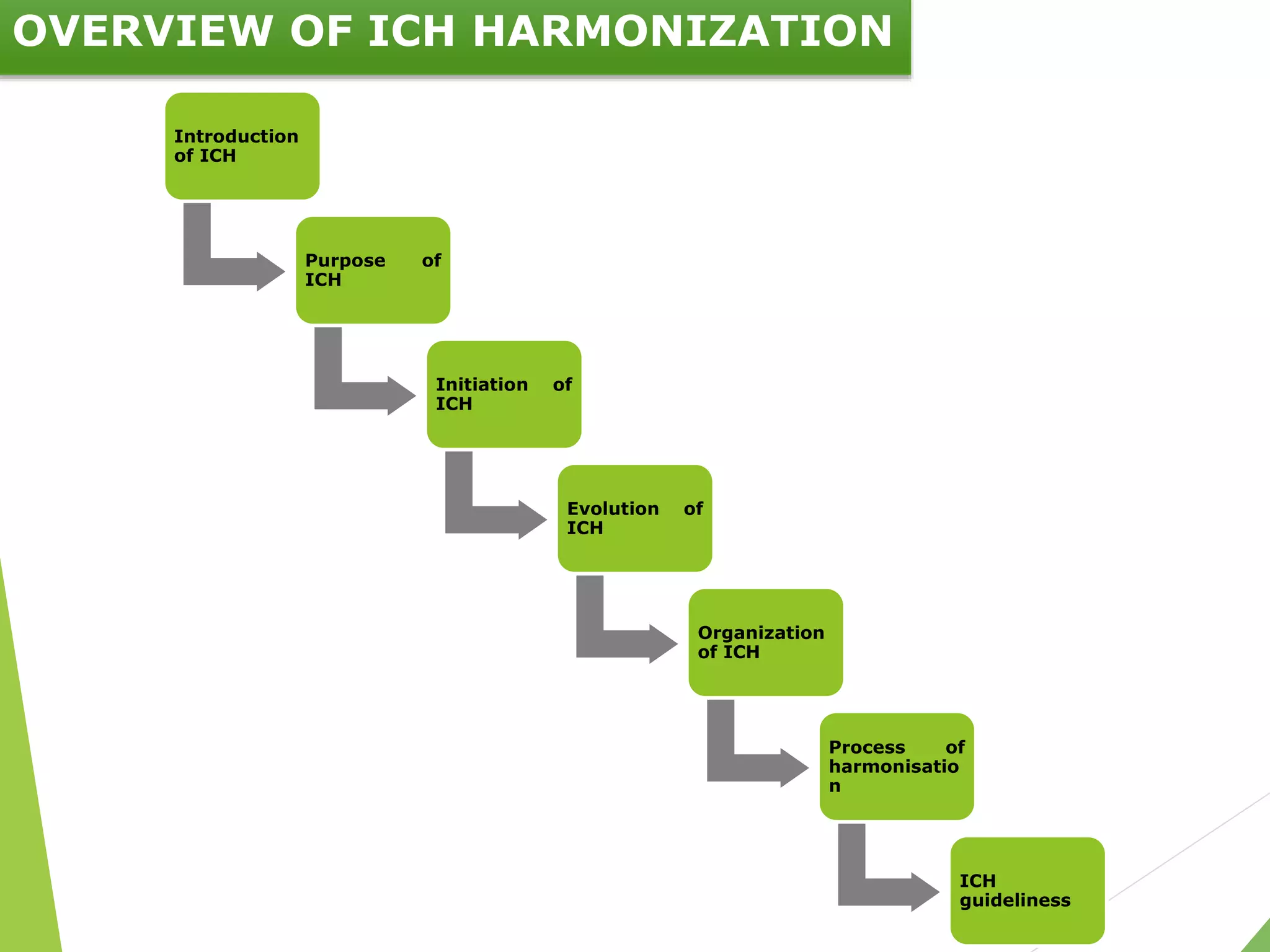














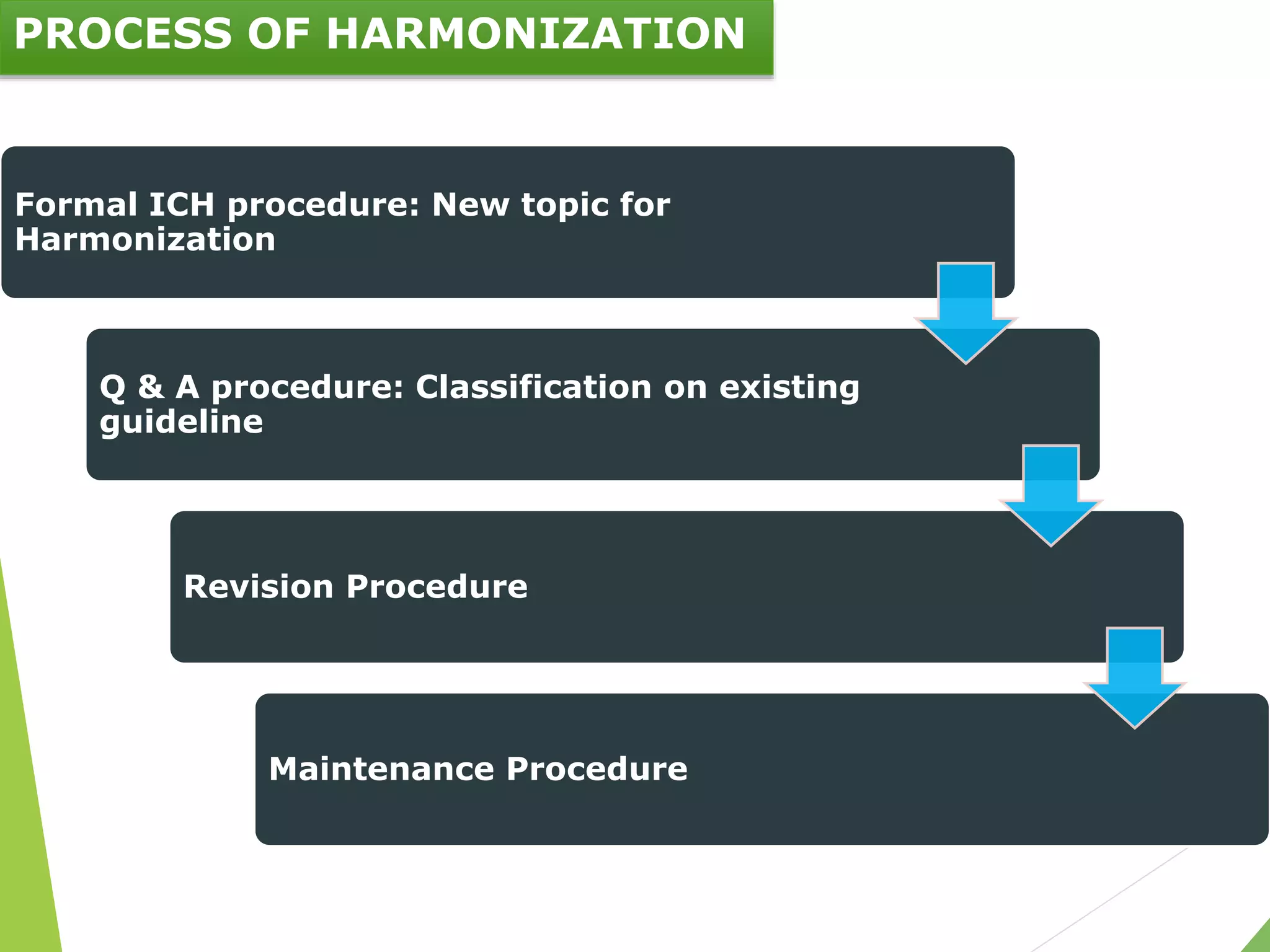
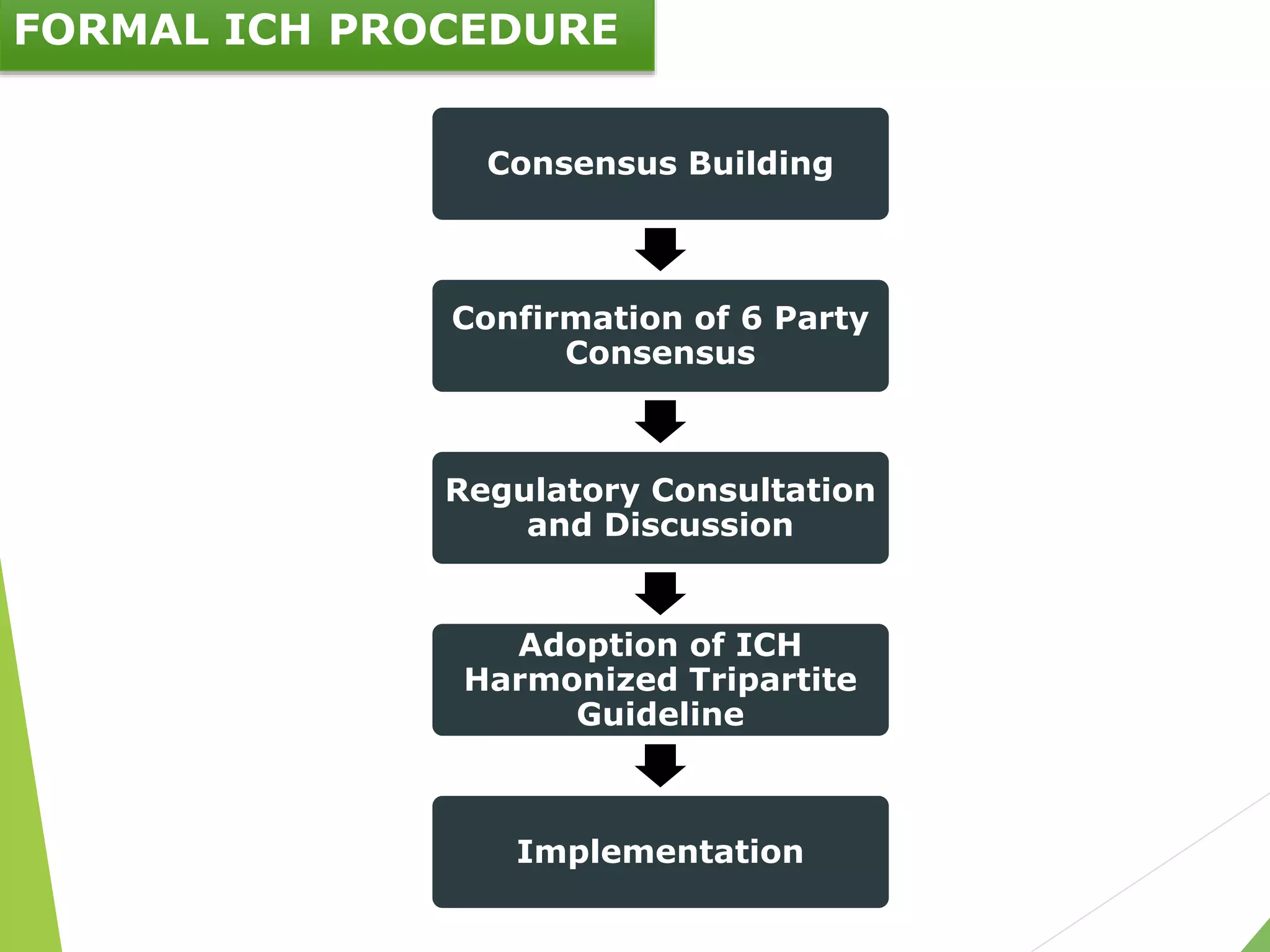














The document presents an overview of the International Council on Harmonisation (ICH), detailing its purpose, evolution, organization, and processes for harmonizing pharmaceutical regulations. Established in 1990, ICH aims to ensure the safety, efficacy, and quality of medicines while minimizing animal testing and clinical trial duplication. The document also outlines the types of guidelines developed by ICH and emphasizes the need for global cooperation in pharmaceutical development and oversight.



































































