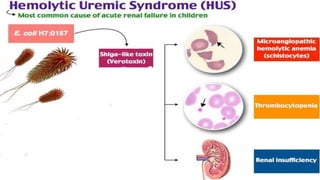
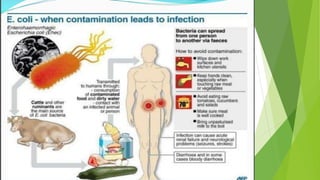
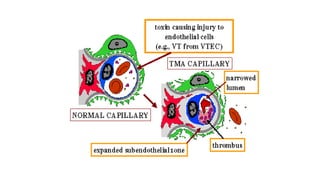
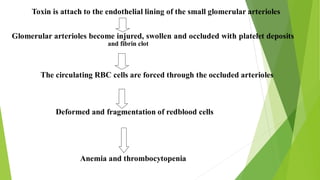
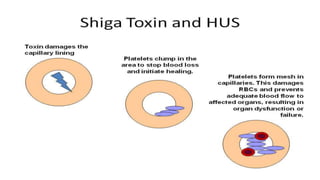
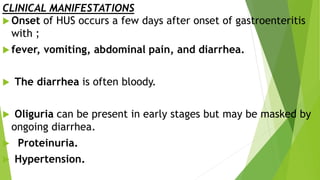
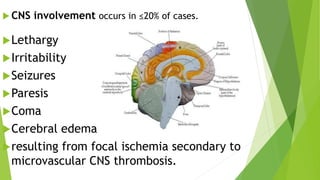
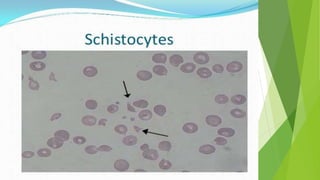
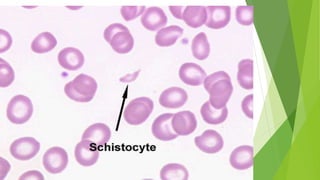
Hemolytic uremic syndrome (HUS) is a disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal injury, most commonly caused by toxin-producing strains of E. coli. The toxins produced by these bacteria damage endothelial cells in the kidneys and other organs, leading to platelet aggregation and thrombosis. This causes fragmentation of red blood cells and kidney dysfunction. HUS diagnosis is based on laboratory findings of hemolytic anemia, low platelet count, and kidney involvement. Treatment is largely supportive through dialysis, blood transfusions, and controlling blood pressure and electrolyte abnormalities. Most patients recover renal function, but some are left with chronic kidney disease.