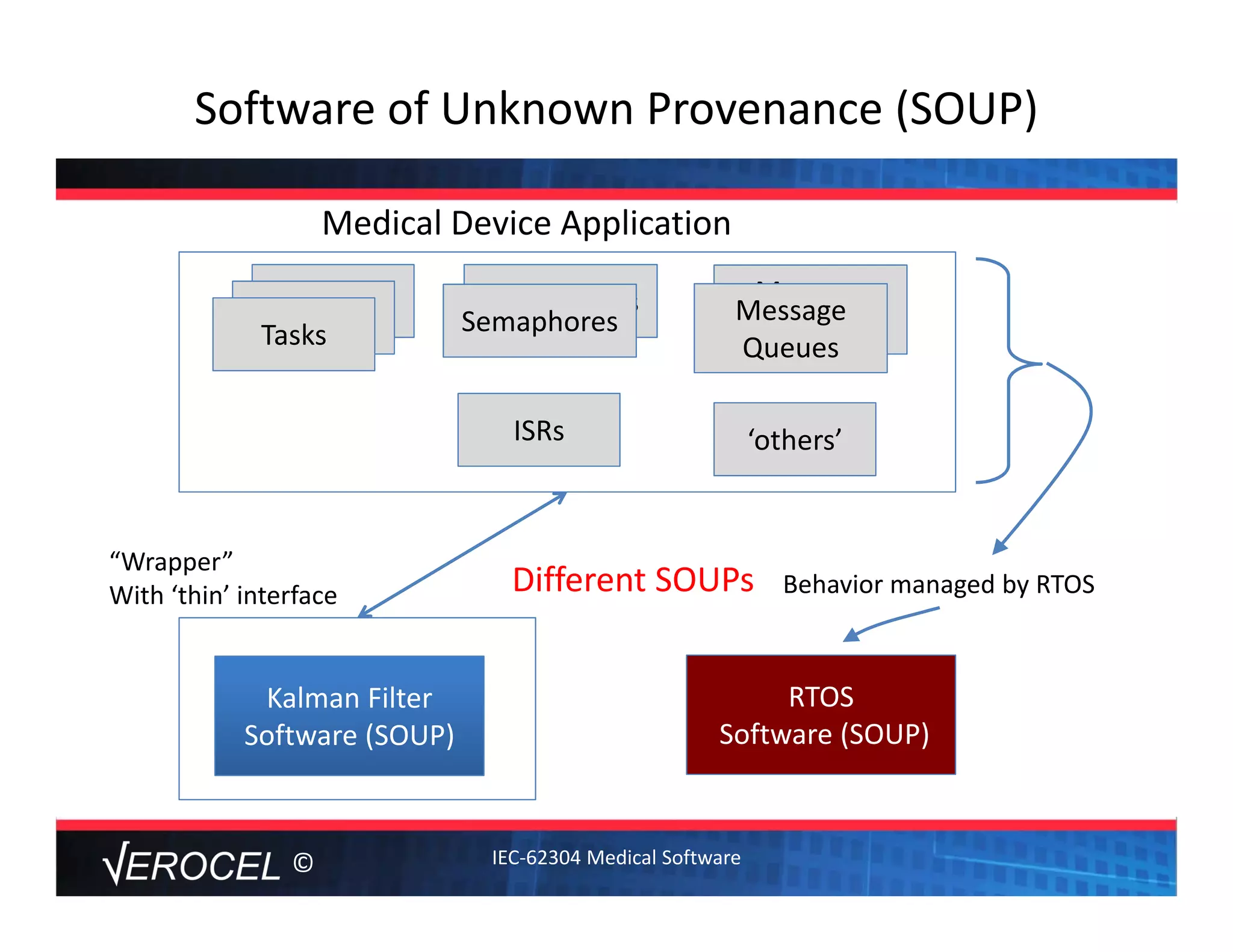
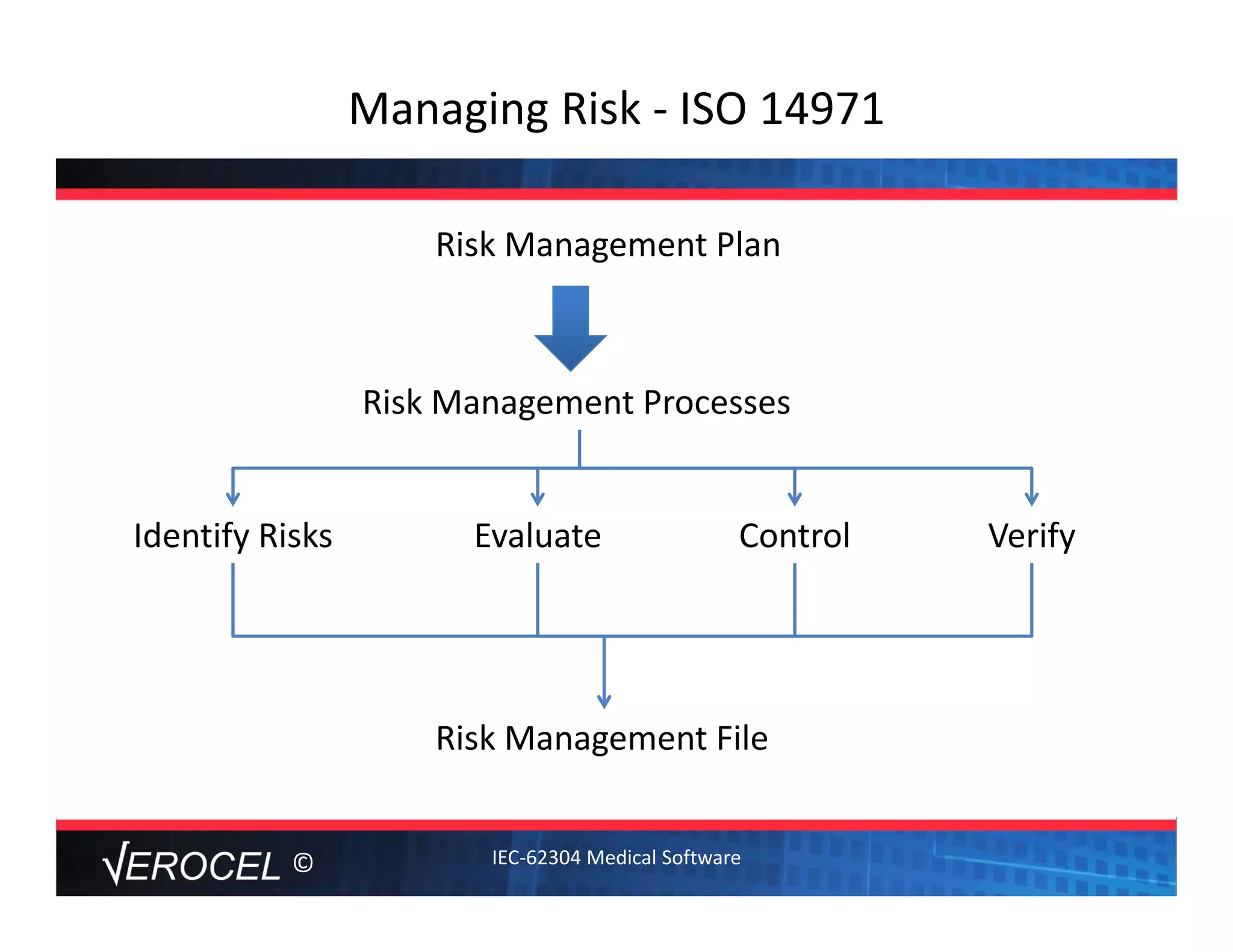
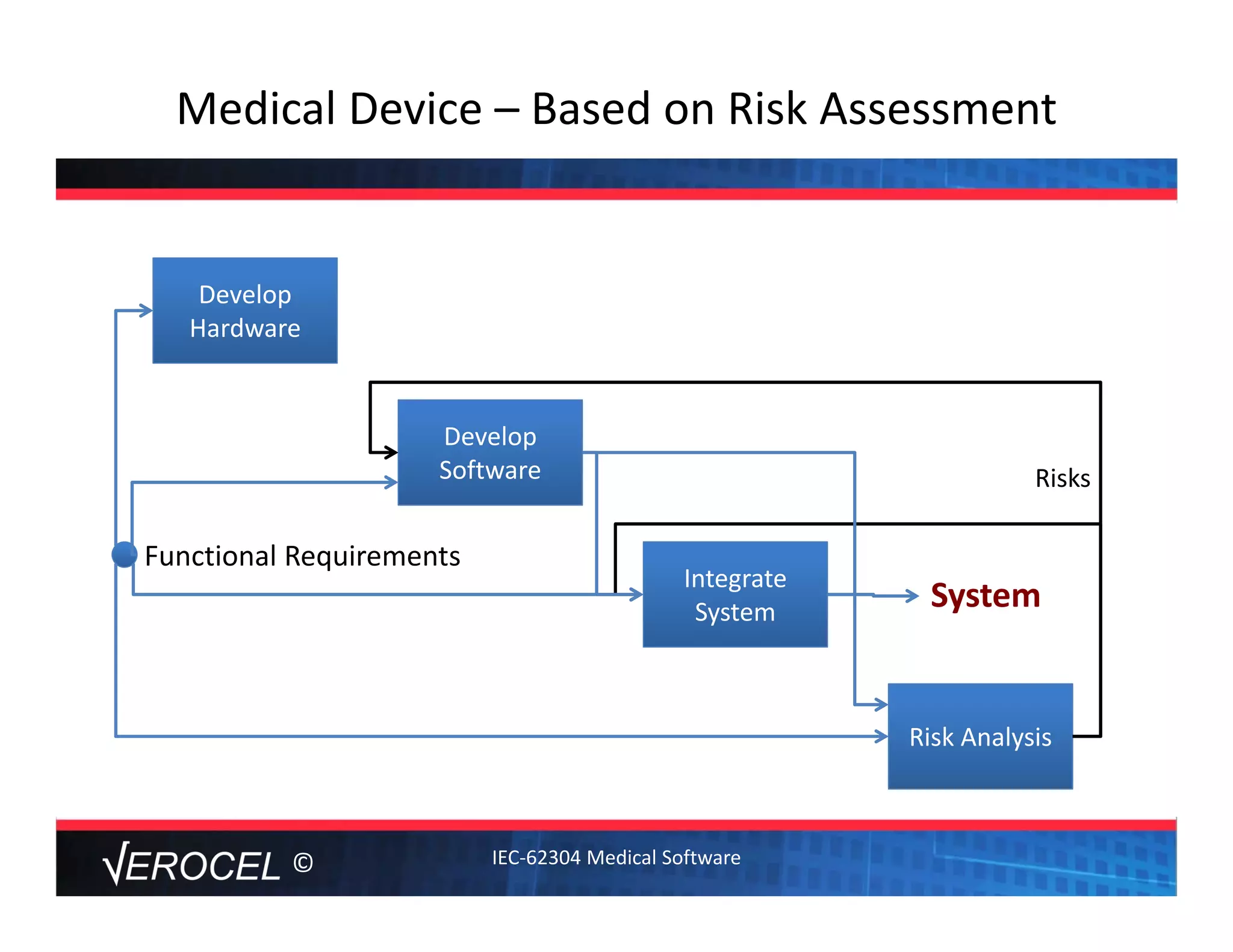
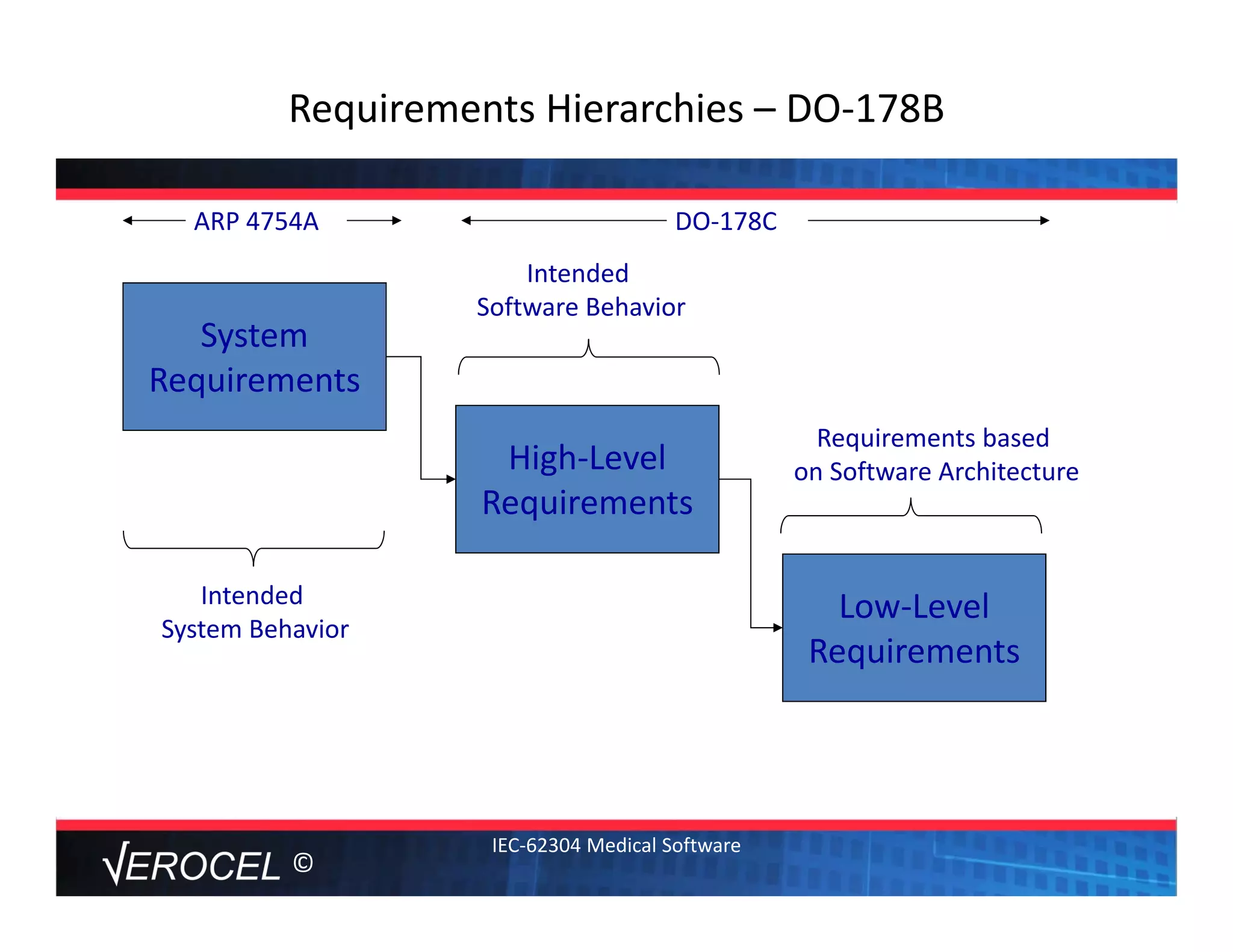
This document summarizes key information from a seminar on getting medical devices approved by the FDA. It discusses common reasons why companies fail FDA approval, including inadequate software documentation. The document outlines FDA guidance and standards for software documentation, design, validation, and human factors. It also discusses FDA concerns about cybersecurity for networked devices and provides an overview of FDA guidance on managing cybersecurity risks.















![Regulatory Basis for Human Factors
21 CFR 820.30, Design Controls (The need for human
factors is implied):
Design input – includes “needs of the user and
patient”
Design verification – performance criteria met
Design validation – “… devices conform to defined
user needs and intended uses and shall include
testing of production units under actual or
simulated use conditions. Design validation shall
include software validation and risk analysis….”
[incl. use‐related risks]
17](https://image.slidesharecdn.com/gettingdevicefdaapproved-140605135731-phpapp01/75/Getting-Your-Medical-Device-FDA-Approved-18-2048.jpg)




































































































![Software design for_medical_devices_europe_conferent_19012011[1]](https://cdn.slidesharecdn.com/ss_thumbnails/softwaredesignformedicaldeviceseuropeconferent190120111-110119154405-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






















![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)







