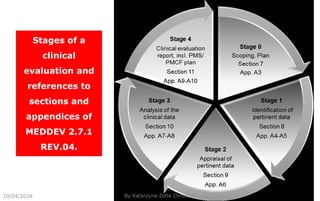
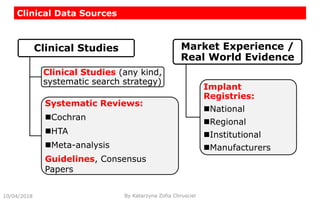
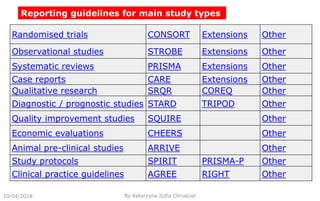
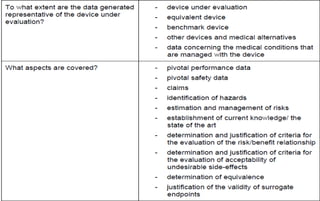
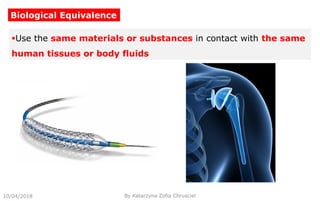
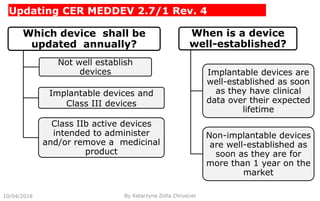
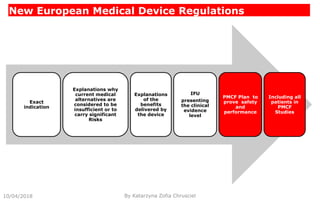
This document summarizes key aspects of clinical evaluation requirements for medical devices in Europe. It discusses the stages of clinical evaluation, including scope definition, identification of pertinent clinical data, appraisal of data, and analysis of clinical data. It notes that clinical evaluation is mandatory for initial CE marking and must be updated. It outlines criteria for determining the quality, relevance and contribution of clinical data sources. Equivalence must be demonstrated when using data from equivalent devices. Clinical investigations are generally required for class III and implantable devices to demonstrate safety and performance.













![Stage 0 – Scope Definition and Clinical Evaluation Plan
Product and related accessories
List of affected products (sizes)
Which requirements are to be fulfilled (e.g. MDD, AIMDD,
[MDR])
Which guidance documents are followed (MEDDEV, Common
Specifications)
List of applicable clinically relevant standards (e.g. ISO 14155)
10/04/2018 By Katarzyna Zofia Chrusciel](https://image.slidesharecdn.com/cer-pms-pmcf-181122153708/85/CER-PMS-PMCF-14-320.jpg)






























































































































![Educo Life Science [gathering clinical evidence] [module 1]](https://cdn.slidesharecdn.com/ss_thumbnails/educo-gatheringclinicalevidence-module1-220128131137-thumbnail.jpg?width=640&height=640&fit=bounds)





























