Downloaded 111 times






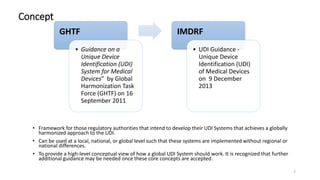
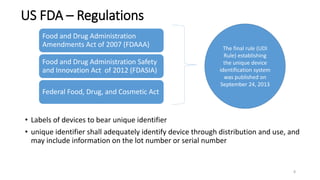
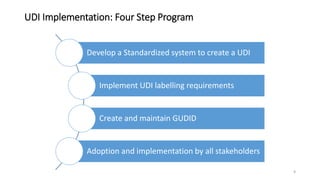



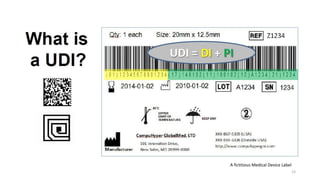







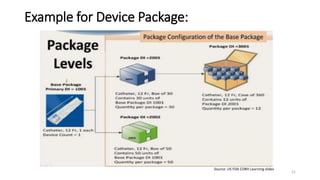






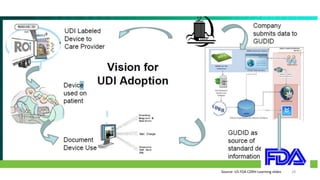
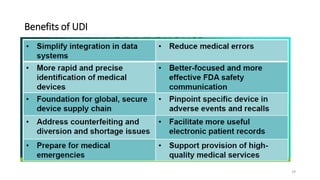



The document provides an overview of unique device identification (UDI) regulations in the United States. It defines key terms related to UDI such as device identifier and production identifier. It explains that UDI is composed of these two identifiers and facilitates rapid identification of medical devices. The document also summarizes UDI requirements including placement of UDI on labels and packages, direct marking requirements, exceptions, and compliance dates. It describes the Global Unique Device Identification Database where UDI information must be submitted.















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)






































