Download as PDF, PPTX

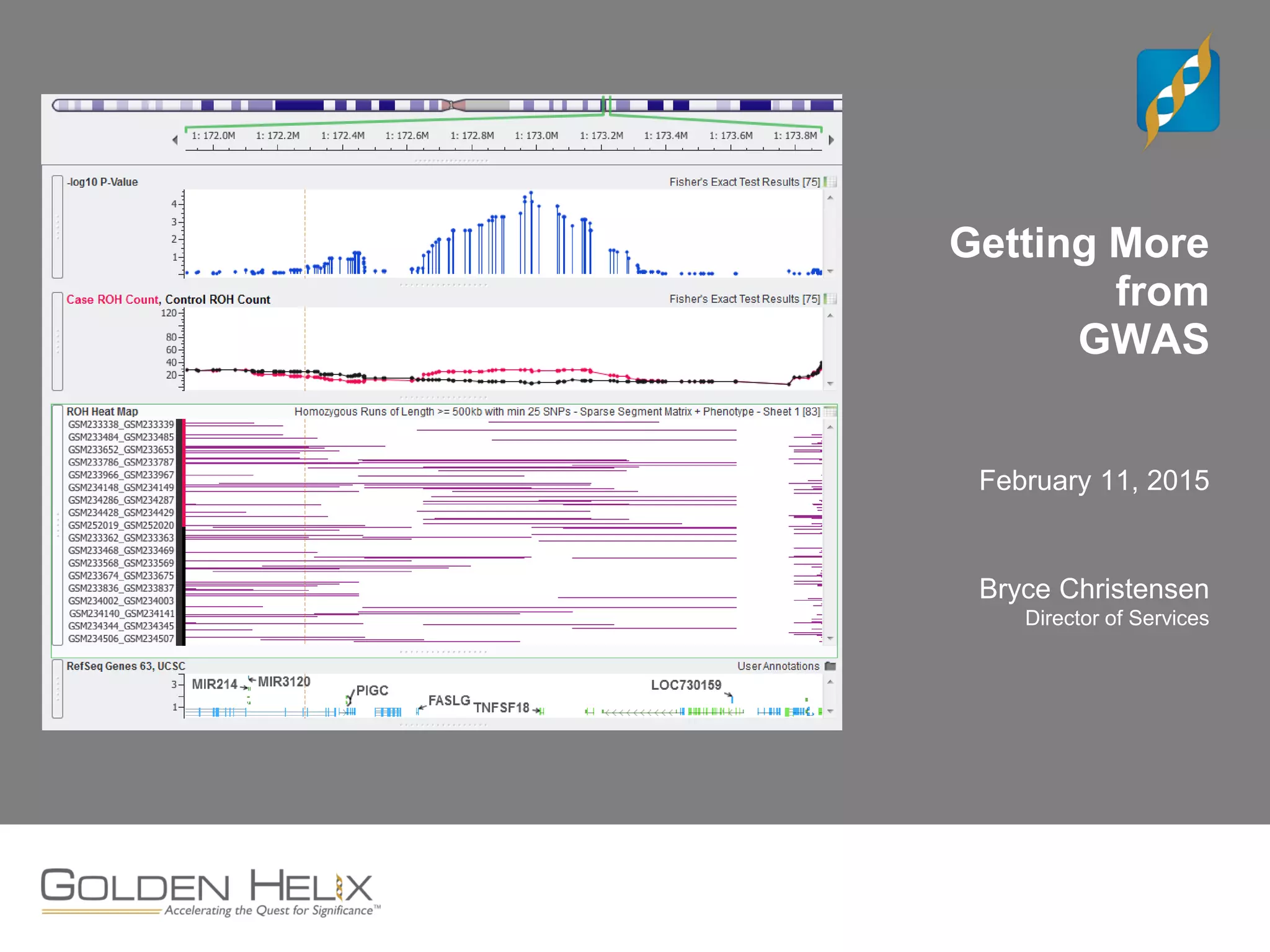



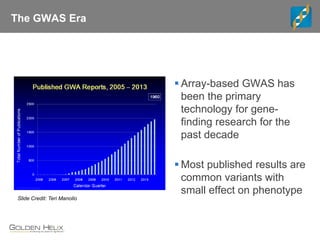
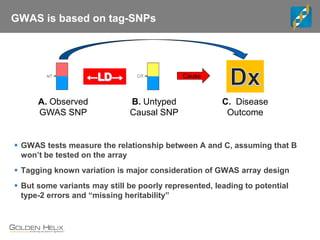





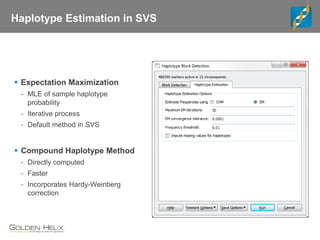
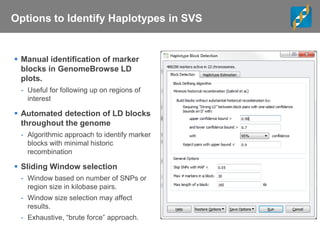


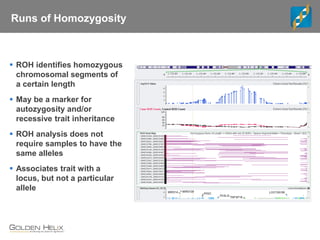
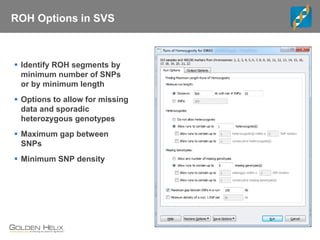




This document discusses ways to get more information from genome-wide association studies (GWAS) data beyond standard single SNP analyses. It describes how haplotype analysis, which considers combinations of neighboring markers, and runs of homozygosity (ROH) analysis, which identifies homozygous chromosomal segments, can reveal genetic associations not evident from individual SNPs. The document demonstrates these techniques using Golden Helix's SNP and Variation Suite software, highlighting options for haplotype phasing, identification and association testing, as well as ROH identification and outputs that could be used to find trait loci.























































































