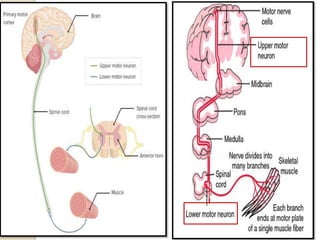
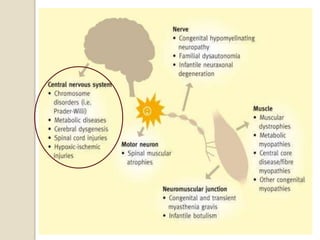
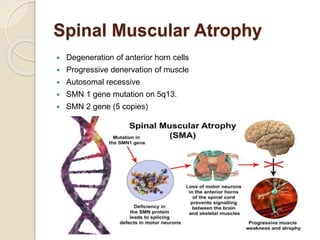
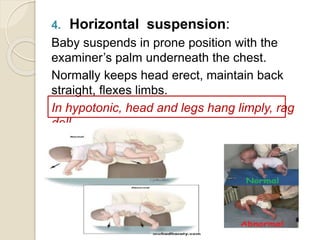
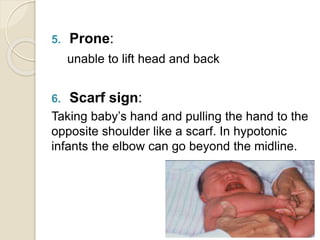
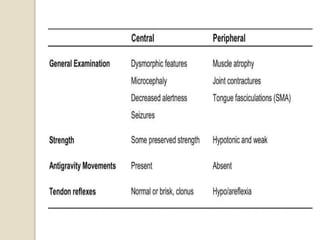
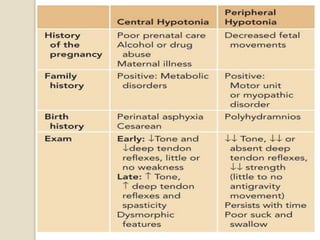
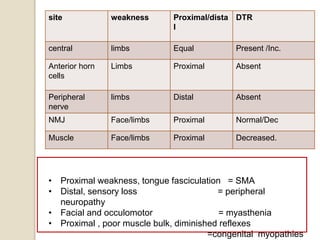
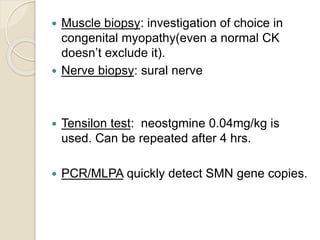
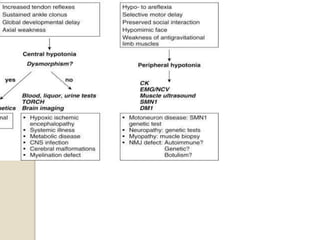
This document discusses the evaluation and management of hypotonia in infants. It begins with an introduction that defines hypotonia, weakness, and the difference between the two. The causes of hypotonia are then explored, separating them into central nervous system causes and peripheral causes. Specific central causes discussed include cerebral insults, metabolic disorders, and benign congenital hypotonia. Peripheral causes mentioned include congenital myopathies, muscular dystrophies, spinal muscular atrophy, and myasthenia gravis. The document outlines the clinical approach for evaluating a hypotonic infant, including taking a thorough history, performing a neurological examination to localize lesions, and determining if the cause is central or peripheral. Key investigations are then reviewed depending on