“H Sanofi δηλώνειρητά και σας ενημερώνει, ότι:
(α) οποιαδήποτε παρουσίαση, συμπεριλαμβανομένου και του συνόλου του υλικού σε
οποιαδήποτε μορφή, το οποίο σχετίζεται με αυτή, έχει αποκλειστικά και μόνο
εκπαιδευτικό χαρακτήρα και σε καμία περίπτωση δεν δύναται να υποκαταστήσει την
επιστημονική σας άποψη, η οποία διαμορφώνεται με ανεξάρτητο τρόπο από εσάς,
(β) To παραπάνω άρθρο βασίζεται στην τρέχουσα ιατρική γνώση και κλινική πρακτική
όπως περιγράφεται στις Διεθνείς Κατευθυντήριες Οδηγίες και σε δημοσιευμένες
κλινικές μελέτες.
Το κάθε φάρμακο θα πρέπει να χρησιμοποιείται σύμφωνα με τις ενδείξεις του και να
ακολουθεί τις οδηγίες που περιλαμβάνονται στην αντίστοιχη Περίληψη
Χαρακτηριστικών του Προϊόντος.”
Disclaimer
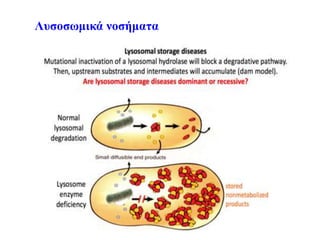
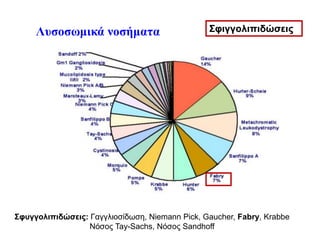
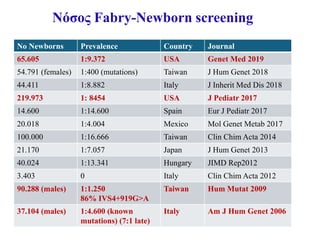
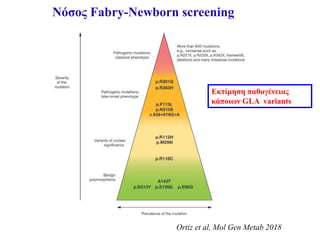
Νόσος Fabry-Newborn screening
•Συστηματικό νεογνικό screening στην Ευρώπη γίνεται μόνο
στην Ιταλία
• Συστηματικό νεογνικός έλεγχος για διάφορα λυσοσωμιακά
νοσήματα στην Taiwan και κάποιες πολιτείες των ΗΠΑ
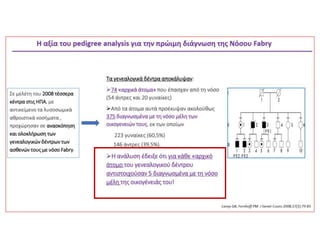
Germain et al, Clinical Genetics 2019
9.
Νόσος Fabry-Αιτιολογία
Προκαλείται από:
•Ανεπάρκεια μερική ή ολική του λυσοσωμικού ενζύμου α-
γαλακτοσιδάση Α (α-galactosidase A: α-Gal A)
• Υπεύθυνο για τον καταβολισμό των γλυκοσφιγγολιπιδίων
(glycosphingolipids)
• Τα γλυκοσφιγγολιπίδια έχουν ένα τελικό άκρο αγαλακτόζης (α-
galactosyl) που είναι συστατικό των περισσότερων κυτταρικών
μεμβρανών (παρεγχυματικών & ενδοθηλιακών)
Brady et al, N Engl J Med 1967
10.
Νόσος Fabry-Αιτιολογία
Συσσώρευση αδιάσπαστωνγλυκοσφιγγολιπιδίων
• Σφαιροτριαοζυλοκεραμίδιο (globotriaosylceramide: Gb3) (το
κύριο) (κυτταροτοξική-προφλεγμονώδη-προϊνώδης δράση)
• Γαλαβιοσυλκεραμίδη (Digalactosylceramide)
• P1 glycolipids
• Γλυκοσφιγγολιπίδιο του αντιγόνου ομάδας B και B1 των
ερυθροκυττάρων
Desnick et al, J Pediatr 2004
Νόσος Fabry-Αιτιολογία
Η συσσώρευσηγίνεται:
Στα λυσοσώματα των περισσότερων κυττάρων του οργανισμού
• ΚΝΣ
• Αυτόνομο νευρικό σύστημα (προσβολή ινών)
• Νωτιαία γάγγλια
• Σε όλα τα νεφρικά κύτταρα (επιθηλιακά κύτταρα του σπειράματος,
ποδοκύτταρα, νεφρικά σωληναριακά επιθηλιακά και μεσαγγειακά
κύτταρα)
• Μυοκαρδιακά κύτταρα
• Ινοβλάστες
• Κύτταρα λύων μυικών ινών
• Ενδοθηλιακά κύτταρα των αγγείων
Desnick et al, J Pediatr 2004
13.
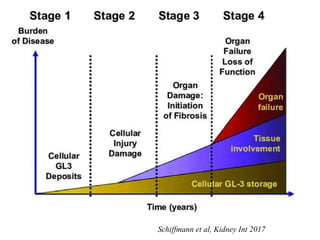
Νόσος Fabry-Αιτιολογία
• Στουςασθενείς με την κλασική μορφή η συνάθροιση στο ενδοθήλιο
των αγγείων οδηγεί σε απόφραξη και ισχαιμία
• Πολυσυστηματικό νόσημα με αγγειακές βλάβες
• Κύρια προσβολή νευρικού συστήματος, δέρματος, καρδιάς,
νεφρών, οφθαλμών
• To νόσημα καταλήγει σε απειλητική για τη ζωή νεφρικές, καρδιακές
και αγγειοεγκεφαλικές εκδηλώσεων
Ries et al, Eur J Pediatr 2003; Desnick et al, J Pediatr 2004
Νόσος Fabry
• ΜΟεπιβίωσης των ασθενών με κλασική νόσο Fabry: περίπου 50 έτη
• ΜΟ επιβίωσης των ασθενών με κλασική νόσο Fabry πριν τη δυνατότητα
εξωνεφρικής κάθαρσης και μεταμόσχευσης: περίπου 40 έτη
MacDermot, J Med Genet 2001
16.
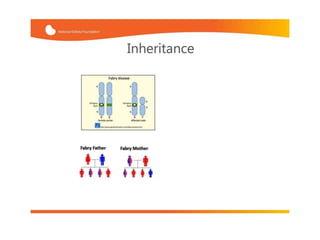
Νόσος Fabry-Αιτιολογία
• X-linkedυπολειπόμενη κληρονομικότητα
• ΟΜΙΜ: 301500
To GaL A γονίδιο (GLA) εδράζεται στο Xq22.1
Bernstein et al, J Clin Invest 1989
18.
Νόσος Fabry-Αιτιολογία
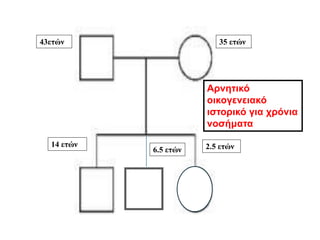
• Ηαπουσία οικογενειακού ιστορικού δεν αποκλείει τη νόσο
Αυτόματες μεταλλάξεις έχουν καταγραφεί
Μεταβίβαση παθολογικού γονιδίου από την μητρική πλευρά, με μια ή
περισσότερες γενιές ασυμπτωματικών ή ήπια προσβεβλημένων
γυναικών
Ashton et al, J Invest Med 2000
19.
• 74 Japaneseοικογένειες με Fabry disease
• 5/74 (6.8%) de novo μετάλλαξη (αρνητικό ιστορικό στους γονείς τους)
Νόσος Fabry-Κληρονομικότητα
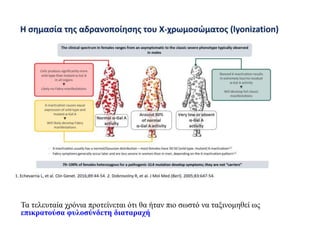
Ετερογένεια στηδιάγνωση λόγω τυχαίας αδρανοποίησης X χρωμοσώματος
oΤο ένα από τα 2 χρωμοσώματα Χ των θηλυκών αδρανοποιείται (~ 16η ήμερα
μετά τη γονιμοποίηση, στάδιο 500-1000 κυττάρων)
oΣωμάτιο Barr: συμπυκνωμένο, αδρανοποιημένο Χ.
oΤο χρωμόσωμα Χ που αδρανοποιείται επιλέγεται τυχαία από τα πατρικής (Π)
και μητρικής (Μ) προέλευσης χρωμοσώματα
oΌταν ένα Π ή Μ χρωμόσωμα αδρανοποιηθεί σε ένα κύτταρο τότε και όλοι οι
απόγονοι αυτού του κυττάρου κληρονομούν το ίδιο πρότυπο αδρανοποίησης
oΤυπικά τα προερχόμενα από τον πατέρα και τη μητέρα χρωμοσώματα Χ
αδρανοποιούνται περίπου με τη ίδια συχνότητα 1:1
oΕξ αιτίας της τυχαίας αδρανοποίησης του χρωμοσώματος Χ, τα φυλοσύνδετα
χαρακτηριστικά εκφράζονται ποικιλόμορφα στις (ετερόζυγες για φυλοσύνδετο
γονίδιο) γυναίκες
22.
Τα τελευταία χρόνιαπροτείνεται ότι θα ήταν πιο σωστό να ταξινομηθεί ως
επικρατούσα φυλοσύνδετη διαταραχή
23.
Νόσος Fabry-Αιτιολογία
• Ανευρίσκεταισε όλες τις εθνότητες
• Έχουν καταγραφεί εκατοντάδες μεταλλάξεις στο γονίδιο α-Gal A
• Οι πολλές μεταλλάξεις ίσως εξηγούν την κλινική ποικιλομορφία της
νόσου
• Οι περισσότερες οικογένειες έχουν αποκλειστικές μεταλλάξεις
• Σημαντικές διαφορές στις εκδηλώσεις της νόσου έχουν βρεθεί σε
ημιζυγώτες στην ίδια οικογένεια
Hasholt et al, J Med Genet 1990; Knol et al, Am J Med Genet 1999
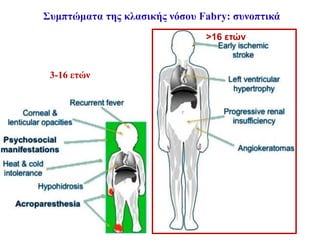
Κλασική νόσος Fabry-Κλινικήεικόνα
• Αγόρια έναρξη συμπτωμάτων: ΜΟ 10.9 έτη
• Κορίτσια έναρξη συμπτωμάτων: ΜΟ 22.6 έτη
• ΜΟ καθυστέρησης της διάγνωσης από τα πρώτα συμπτώματα:
αγόρια 13.7 έτη
κορίτσια 16.3 έτη
Morgan and Crawfurd, BMJ 1988; Miekle et al, JAMA 1999;
Mehta et al, Eur J Clin Invest 2004
27.
Κλασική νόσος Fabry-Κλινικήεικόνα
• Αντίθετα με τα υπόλοιπα λυσοσωμικά νοσήματα, η νόσος του Fabry
δεν σχετίζεται με νοητική υστέρηση
• Παρόλο που τα βασικά συμπτώματα εμφανίζονται στην παιδική
ηλικία συχνά αυτά παραβλέπονται
• Τα συμπτώματα στην παιδική ηλικία μπορεί να είναι ποικίλα και
ύπουλα
Morgan and Crawfurd, BMJ 1988; Miekle et al, JAMA 1999
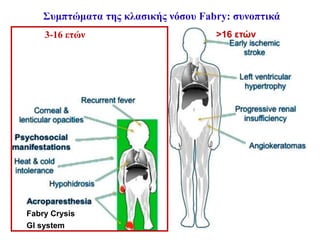
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Κρίσεις Fabry (Οξέα επεισόδια πόνου)
• Το πιο εντυπωσιακό σύμπτωμα-«Δραματικό νευροπαθητικό
άλγος»
Εμφανίζονται στα περισσότερα αγόρια με κλασική νόσο
Έναρξη από παλάμες και πέλματα και επέκταση κεντρικότερα
Διάρκεια από λεπτά έως εβδομάδες
MacDermot et al, J Med Genet 2001; Brady and Schiffmann, JAMA 2000
31.
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Κρίσεις Fabry (Οξέα επεισόδια πόνου)
• Συχνά εκλύεται από stress, άσκηση, κόπωση, ζέστη
• Σχετίζονται με την υποϊδρωσία και την αδυναμία διατήρησης της
θερμοκρασίας του σώματος μέσω της εφίδρωσης, οδηγώντας έτσι σε
αύξηση της εν τω βάθει θερμοκρασίας, η οποία οδηγεί στο άλγος
• Η υποϊδρωσία οδηγεί επίσης σε δυσανεξία στη ζέστη και άσκηση, η
οποία επιδεινώνεται με την ηλικία
MacDermot et al, J Med Genet 2001; Brady and Schiffmann, JAMA 2000
32.
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Ακροπαραισθησία:
Δεύτερος τύπος άλγους λόγω περιφερικής νευροπάθειας
Εμφάνιση και στις γυναίκες φορείς στην εφηβεία
Χρόνιο καυστικό και κνησμώδες άλγος σε παλάμες & πέλματα
Μπορεί να είναι καθημερινό, διάρκειας από λεπτά έως ημέρες
Στα παιδιά συχνά θεωρείται ψυχογενές ή άλγος ανάπτυξης
Οι συχνές υποτροπές και η αδυναμία ιατρικής εξήγησης συχνά οδηγούν σε
κατάθλιψη στην εφηβεία
Filling-Katz et al, Neurology 1989; MacDermot et al, J Med Genet 2001
Έναρξη στα ♂: 3 έτη, ΜΟ 10 έτη
♀: 6 έτη, ΜΟ 15 έτη
33.
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Ακροπαραισθησία
• Η φυσική εξέταση συνήθως είναι φυσιολογική
• Το ΗΜΓ και οι ταχύτητες αγωγής συνήθως είναι φυσιολογικά, διότι
αρχικά προσβάλλονται μικρές νευρικές ίνες
34.
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Συμπτώματα από το γαστρεντερικό σύστημα
Έναρξη στην παιδική ηλικία
Ήπιας έως βαριάς μορφής
Έναρξη στην εφηβεία, νεαρή ενήλικη ζωή Κορίτσια φορείς
Διάρροια, ναυτία, έμετοι, δυσκοιλιότητα
Ανεπαρκής πρόσληψη ΒΣ
Οξύ κοιλιακό άλγος (μιμείται τη σκωλεικοειδίτιδα)
Sheth et al, Am J Gastroenterol 1981; Argoff et al, Nucl Med Commun 1998
αγόρια
35.
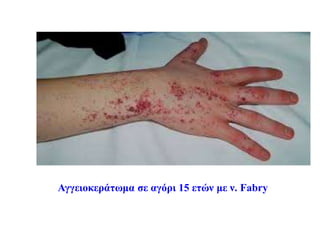
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
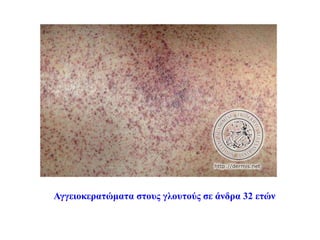
Δερματολογικές εκδηλώσεις
• Σχεδόν όλοι οι άρρενες ασθενείς έχουν αγγειοκερατώματα
• Το 10-35% των θηλέων φορέων στην εφηβεία εμφανίζουν
αγγειοκερατώματα, πιο περιορισμένα (κηλίδες θώρακα, βουβωνικής
χώρας, οσφύος)
• Αυξάνονται σε αριθμό και μέγεθος με την ηλικία
• Μικρές, ελαφρώς προεξέχουσες βαθυκόκκινες βλάβες (purplish-red,
nonbranching telangiectases)
• Μεταξύ ομφαλού και γονάτων, αλλά και σε άλλες θέσεις (πχ
βλεννογόνος στόματος)
Brady and Schiffmann, JAMA 2000
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Θολερότητα κερατοειδούς
• Τα περισσότερα αγόρια, >70% των ετερόζυγων θήλεων
• Δεν επηρεάζεται η όραση
MacDermot et al, J Med Genet 2001
41.
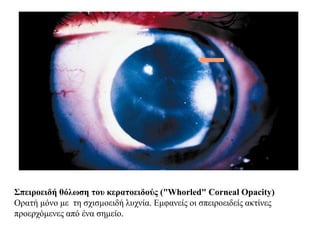
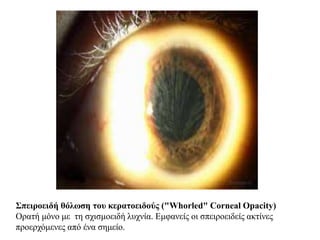
Σπειροειδή θόλωση τουκερατοειδούς ("Whorled" Corneal Opacity)
Ορατή μόνο με τη σχισμοειδή λυχνία. Εμφανείς οι σπειροειδείς ακτίνες
προερχόμενες από ένα σημείο.
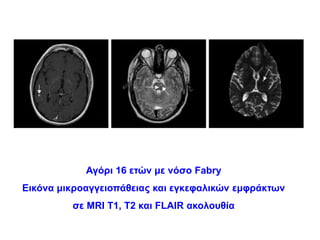
Αγόρι 16 ετώνμε νόσο Fabry
Εικόνα μικροαγγειοπάθειας και εγκεφαλικών εμφράκτων
σε MRI T1, T2 και FLAIR ακολουθία
45.
Συμπτώματα της κλασικήςμορφής της νόσου Fabry
Όψιμη εφηβεία-ενήλικη ζωή
• Καρδιαγγειακή δυσλειτουργία
* ΜΟ έναρξης μετά τη 2η δεκαετία, ενίοτε σε ηλικία < 20 ετών (♂,♀)
* Έμφραγμα μυοκαρδίου, υπερτροφία αριστερής κοιλίας, ΚΑ
* Βαλβιδοπάθειες (ανεπάρκεια ΜΒ)
* Αρρυθμίες, διαταραχές αγωγιμότητας
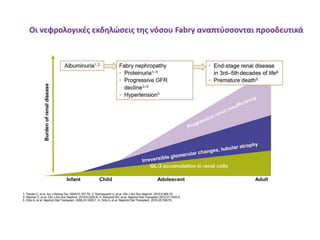
• Νεφρική δυσλειτουργία
* ΜΟ έναρξης μετά τη 2η δεκαετία, ενίοτε σε ηλικία < 20 ετών (♂,♀)
* Επιδείνωση από την 3η ή 4η δεκαετία ζωής
* Ουραιμία, ΑΥ, τελικού σταδίου ΧΝΑ, παραπυελικές κύστεις
Obrador et al, J Am Soc Nephrol 2002; Ries et al, Eur J Pediatr 2003;
Desnick et al, J Pediatr 2004; Ries et al, Kidney Int 2004
Συμπτώματα κλασικής μορφήςτης νόσου
Fabry
Σκελετικές ανωμαλίες
• Τα παιδιά με νόσο Fabry δεν εμφανίζουν τις σκελετικές
ανωμαλίες που χαρακτηρίζουν άλλα λυσοσωμικά
νοσήματα
• Καθυστέρηση αύξησης και εφηβείας
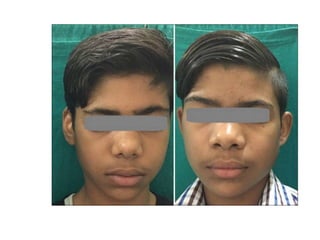
• Αραιές, λεπτές τρίχες στο πρόσωπο και στο σώμα
• Χαρακτηριστική δυσμορφία του προσώπου >50% των
αρρένων, αναγνωρίσιμη στην πρώιμη εφηβεία
MacDermot et al, J Med Genet 2001
50.
Ries et al,Genet Med 2006
Facial images of patients with Fabry disease with frontal face directly above
profile view of the same patient arranged from youngest to oldest. χ denotes
cousins.
Διαγνωστική προσέγγιση τηςνόσου Fabry
Αγόρια
Ανεπάρκεια ή απουσία α-Gal A δραστηριότητας στο πλάσμα,
λευκοκύτταρα του περιφερικού αίματος, δάκρυα, βιοψία ιστού. Οι
φυσιολογικές τιμές ποικίλουν στα διάφορα εργαστήρια.
Κορίτσια
Στα θήλεα άτομα, πολύ χαμηλά επίπεδα α-Gal A δραστηριότητας είναι
επίσης διαγνωστικά για τα άτομα φορείς
Ωστόσο, φυσιολογικά ή σχεδόν φυσιολογικά επίπεδα α-Gal A
δραστηριότητας δεν αποκλείουν ένα θήλυ να είναι φορέας
Οι ετεροζυγώτες γυναίκες μπορεί να έχουν φυσιολογικά επίπεδα α-Gal A
δραστηριότητας λόγω τυχαίας αδρανοποίησης του Χ χρωμοσώματος
Όλα τα κορίτσια και οι γυναίκες υψηλού κινδύνου πρέπει να υποβάλλονται
σε γενετική μελέτη για την ανίχνευση της μετάλλαξης
Desnick et al, J Lab Clin Med 1973
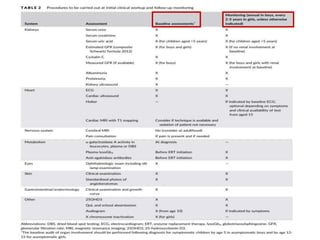
Νόσος Fabry-Θεραπεία
• Χρειάζεταισυνεργασία παιδιάτρου και ιατρών άλλων ειδικοτήτων
(οφθαλμίατρου, νευρολόγου, γενετιστή, καρδιολόγου,
ρευματολόγου και δερματολόγου)
55.
Θεραπεία νόσου Fabry
•Υποστηρικτική θεραπεία του άλγους, των καρδιακών,
εγκεφαλοαγγειακών επιπλοκών και της ΧΝΑ με εξωνεφρική
κάθαρση
• Αύξηση της επιβίωσης
Ενζυμική θεραπεία νόσουFabry
• Η παθολογία της νόσου Fabry ξεκινά από τη γέννηση ή και πριν τη
γέννηση, παρά την ήπια αρχικά εικόνα
• Η ενζυμική θεραπεία υποκατάστασης παρέχει στους ασθενείς την
πρωτεΐνη που ανεπαρκεί και διαφυλάσσει από τις μεταβολικές
ανωμαλίες
Elleder et al, Cesk Patol 1998; Schiffmann et al, Proc Natrl Acad Sci USA 2000;
Moore et al, BMC Neurol 2002
58.
Ενζυμική θεραπεία νόσουFabry
• Ανασυνδυασμένες μορφές ανθρώπινου ενζύμου α-Gal A
1. Αγαλσιδάση Α/0.2 mg/Kg ΙV έγχυση (κάθε 2η εβδομάδα)
2. Αγαλσιδάση Β/1 mg/Kg ΙV έγχυση (κάθε 2η εβδομάδα)
• Πολλές δομικές και λειτουργικές ομοιότητες
μειώνει τη συσσώρευση Gb3 σε πολλούς τύπους κυττάρων,
συμπεριλαμβανομένων ενδοθηλιακών και παρεγχυματικών
Linthorst et al, J Inher Metab Dis 2002; Lee et al, Glycobiology 2003
59.
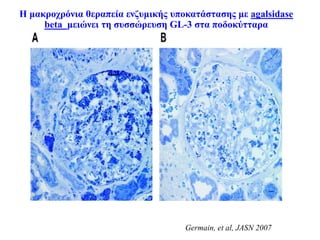
H μακροχρόνια θεραπείαενζυμικής υποκατάστασης με agalsidase
beta μειώνει τη συσσώρευση GL-3 στα ποδοκύτταρα
Germain, et al, JASN 2007
60.
Ενζυμική θεραπεία νόσουFabry
Σημαντική η πρώιμη έναρξη αγωγής
προς αποφυγή μόνιμων βλαβών
Μελέτες έχουν δείξει ότι η πρώιμη έναρξη
α-Gal A θεραπεία υποκατάστασης μπορεί να εμποδίσει την εμφάνιση
της νόσου ή να αποτρέψει την εξέλιξη
Schiffmann et al, Proc Natl Acad Sci USA 2000; Moore et al BMC Neurol 2002;
Germain et al, Clinical Genetics 2019
61.
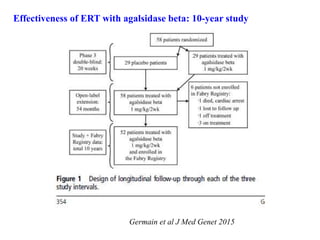
Effectiveness of ERTwith agalsidase beta: 10-year study
Germain et al J Med Genet 2015
62.
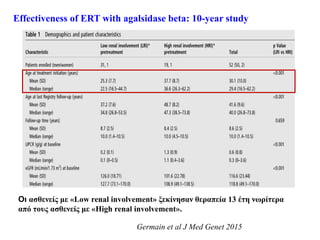
Effectiveness of ERTwith agalsidase beta: 10-year study
Germain et al J Med Genet 2015
Low renal involvement
UPCR <0,5 g/g και <50%
σκληρυμένα σπειράματα
High renal involvement
UPCR >0,5 g/g και >50%
σκληρυμένα σπειράματα
63.
Οι ασθενείς με«Low renal involvement» ξεκίνησαν θεραπεία 13 έτη νωρίτερα
από τους ασθενείς με «High renal involvement».
Effectiveness of ERT with agalsidase beta: 10-year study
Germain et al J Med Genet 2015
64.
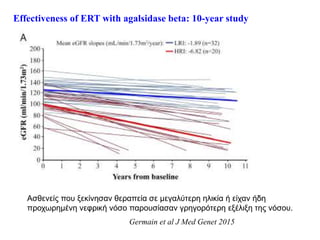
Effectiveness of ERTwith agalsidase beta: 10-year study
Germain et al J Med Genet 2015
Ασθενείς που ξεκίνησαν θεραπεία σε μεγαλύτερη ηλικία ή είχαν ήδη
προχωρημένη νεφρική νόσο παρουσίασαν γρηγορότερη εξέλιξη της νόσου.
65.
• Συμπτωματικά αγόριακαι κορίτσια
νευροπαθητικό άλγος
παθολογική αλβουμινουρία (≥3 mg/mmol ή ≥0.26 mg/mg)
σοβαρή συμμετοχή από το ΓΝΣ και κοιλιακό άλγος
καρδιακή συμμετοχή
Όχι ένδειξη επί του παρόντος για ERT με παρουσία μόνο αγγειοκερατώματος
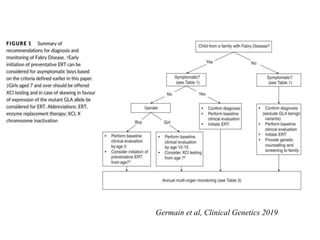
Ενζυμική θεραπεία νόσου Fabry (ΕRT)
Clinical Genetics 2019
66.
• Ασυμπτωματικά αγόρια≥7-8 ετών (απουσία δεδομένων για
πρωιμότερη έναρξη) που έχουν τα ακόλουθα κριτήρια:
παθογόνο Gla variant για classic Fabry
οικογενειακό ιστορικό σοβαρής νόσου στα άρρενα
μη ανιχνεύσιμη ΑGAL-A δραστηριότητα στα περιφερικά λευκοκύτταρα
και lysoGb3 στο πλάσμα >20 nmol/L
• Ασυμπτωματικά κορίτσια όχι δεδομένα για πρώιμη έναρξη ERT
κορίτσια ≥ 7 ετών πρέπει να κάνουν XCl testing και εάν υπάρχει μεγάλη
έκφραση του μεταλλαγμένου γονιδίου, έναρξη ERT
Ενζυμική θεραπεία νόσου Fabry (ΕRT)
Clinical Genetics 2019
67.
Ενζυμική θεραπεία νόσουFabry (ΕRT)
Συστάσεις ειδικών για παρακολούθηση κατά τη διάρκεια της ERT για:
• Αντιδράσεις στην έγχυση
• Ανάπτυξη ΙgG αντισωμάτων τόσο στην α-, όσο και η β-αγαλσιδάση στην
πλειονότητα των αρρένων ασθενών με κλασική νόσο Fabry
ανάπτυξη συνήθως εντός 3 μηνών μετά την 1η έγχυση
με την πάροδο του χρόνου: 40% τάση μείωσης, 14% ανοχή (μη
ανιχνεύσιμα), 35% σταθεροποίηση του τίτλου
έλεγχος θε 6 μήνες
Linthorst GE et al, 2005
Lenders et al Orphanet Journal of Rare Diseases 2018
Άλλες θεραπείες νόσουFabry
Oral Chaperon therapy (μιγαλαστάτη)
• 313 μεταλλάξεις έχουν βρεθεί ως επιδεκτικές θεραπείας, δηλαδή 35-
50% του πληθυσμού Fabry
• Ανευρίσκονται σε ειδικό site
• Δε δουλεύει σε άλλες μεταλλάξεις
• Η μιγαλαστάτη ενδείκνυται για τη μακροχρόνια θεραπεία ενηλίκων και
εφήβων ηλικίας ≥18 ετών (ή ≥ 12 για την Ευρώπη) με Fabry και
μετάλλαξη επιδεκτική θεραπείας
www.galafoldamenabilitytable.com
Mauer et al, UpToDate 2024
72.
• 1η γονιδιακήθεραπεία σε άνθρωπο στον Καναδά το 2017
• 5 ενήλικες με κλασική Fabry disease
• 29-48 ετών
• Φυσιολογική παραγωγή Α-GAL A σε μία εβδομάδα
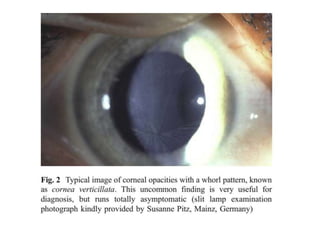
Σπειροειδή θόλωση τουκερατοειδούς ("Whorled" Corneal Opacity)
Ορατή μόνο με τη σχισμοειδή λυχνία. Εμφανείς οι σπειροειδείς ακτίνες
προερχόμενες από ένα σημείο.
Νόσος Fabry-Σύνοψη
• Σημαντικές& παραπλανητικές εκδηλώσεις
• Η πρώιμη διάγνωση είναι σημαντική, υπό το φως της ενζυμικής
θεραπείας υποκατάστασης, η οποία εμποδίζει ή και αναστρέφει
απειλητικές για τη ζωή εκδηλώσεις του νοσήματος
• Οι παιδίατροι πρέπει να είναι υποψιασμένοι σε περιπτώσεις:
σημαντικού, ανεξήγητου πόνου
γαστρεντερικών συμπτωμάτων
υποϊδρωσίας
αγγειοκερατωμάτων
θόλωση του κερατοειδούς
ανεξήγητης υπερτροφίας αρ. κοιλίας
ήπιας πρωτεϊνουρίας & παθολογικών ευρημάτων
γενικής ούρων
85.
Νόσος Fabry
• Σπάνιονόσημα
• Σημαντική η καταγραφή των ασθενών
• Χρήσιμες ιστοσελίδες: Fabrycommunity.com
Fabrazyme.com
Fabryregistry.com
"Rare diseases are rare, but rare disease patients are
numerous” www.orpha.net
86.
Πίσω από κάθε«αναπάντεχο» Fabry σε μια οικογένεια
«κρύβεται» συνήθως μια γυναίκα φορέας….
Ψάξτε την….
«Ακολουθήστε» την…. Εντόπιση και άλλων ασθενών
Ευχαριστώ πολύ
#11 Εκτός από τα αδιάσπαστα γλυκοσφιγγολιπίδια τα οποία συσσωρεύονται στους ασθενείς με νόσο Fabry έχει περιγράφει μια ακόμη ομάδα τα οποία αποτελούν ιδιαίτερα δομικά συστατικά των ερυθροκυττάρων. Υπάρχουν δύο γλυκοσφιγγολιπίδια με την χαρακτηριστική απόληξη a-D-γαλακτοζίδιου, το γλυκοσφιγγολιπίδιο της ομάδας αίματος Β [Gal(al—>3)Gal(2^1aFuc)(pi->3) CIcNAc(Pl->3) G aI(Pl-»4)G Ic(pi^r)C er] το οποίο αναστέλλει την ειδική αιμοσυγκόλληση της ομάδας Β και το γλυκοσφιγγολιπίδιο της ομάδας αίματος Β1 [Gal(al->3)Gai(2<-laFuc)(pi->4)CIcNAc(pl->3)Gal(pl->4)Glc(pi->r)Cer]. Έχει περιγράφει ιδιαίτερα αυξημένη συσσώρευση αυτών στο πάγκρεας ασθενών με νόσο Fabry ενώ τα χαρακτηριστικά και αυτών των λιπιδίων δεν διαφέρουν από αυτά των υγιών ατόμων. Έτσι ασθενείς Fabry με ομάδα αίματος Β και ΑΒ συσσωρεύουν τέσσερα διαφορετικά είδη γλυκοσφιγγολιπιδίων.
#31 Συχνά συνοδεύονται από πυρετό και αυξημένη ΤΚΕ
#36 Τα αγγειοκερατώματα είναι υπερκερατωσικές αγγειακού τύπου βλάβες, και χωρίζονται σε δυο μεγάλες κατηγορίες: τα εντοπισμένα και τα διάχυτα.
Η νόσος Fabry έχει διάχυτο αγγειοκεράτωμα (αγγειοκεράτωμα corporis diffusum)
Εντοπισμένα είναι το αγγειοκεράτωμα Mibelli κληρονομικό (ΑΕ) που εμφανίζεται σε ηλικία 10-14 ετών και
αγγειοκεράτωμα circumscriptum εντοπισμένο που είναι παρόν από τη γέννηση.
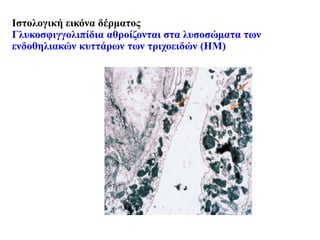
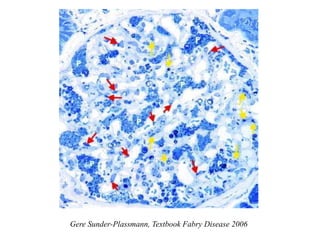
#48 Globotriaosylceramide accumulates in many cell types in the renal glomerulus as dark blue granules and scroll-like whorls, as shown here. Red arrows indicate endothelial accumulation; yellow arrows indicate mesangial cell accumulation; P, indicates podocyte accumulation (magnification, ×40 objective).
#54 CENTOGENE ROSTOCK GERMANY
Η υπηρεσία ForHealth αναλαμβάνει τα προκαταρτικά βήματα αρχικά και υποστηρίζει και στην συνέχεια
Ελληνικό εργαστήριο Genekor με το οποίο συνεργάζεται η Sanofi και διενεργεί δωρεάν 3 panels γονιδίων με ΝGS: για μυοκαρδιοπάθειες (συμπεριλαμβάνει και Fabry, μυοπάθιες (συμπριλαμβάνει και Pompe) και κληρονομικά νοσήματα των νεφρών (συμπεριλαμβάνει και Fabry)
#57 Η μετοκλοπραμίδη (Primperan) είναι φαρμακευτική αντιεμετική ουσία που διεγείρει την κινητικότητα του γαστρεντερικού συστήματος (ΓΕΣ).Η δράση της εντοπίζεται στο ανώτερο τμήμα του ΓΕΣ όπου αυξάνει τον τόνο του οισοφαγικού σφιγκτήρα και διεγείρει τη σύσπαση του πυλωρικού άντρου και του λεπτού εντέρου.
Γκαμπαπεντίνης (Neurontin)
Διφαινυλυδαντοϊνη (Epanutin)
#59 Η αγαλσιδάση άλφα (agalsidase alfa) καταλύει την υδρόλυση του Gb3 (globotriaosylceramide), αποκολλώντας ένα ακραίο κλάσμα γαλακτόζης από το μόριο. Η αγωγή με το ένζυμο αυτό έχει καταδειχτεί ότι μειώνει τη συσσώρευση Gb3 σε πολλούς τύπους κυττάρων, συμπεριλαμβανομένων των ενδοθηλιακών και παρεγχυματικών κυττάρων (Γαληνός)
Η αγαλσιδάση βήτα αποτελεί ανασυνδυασμένη μορφή της ανθρώπινης α-γαλακτοσιδάσης A και παράγεται με εφαρμογή τεχνολογίας ανασυνδυασμένου DNA χρησιμοποιώντας κυτταρική καλλιέργεια θηλαστικού και συγκεκριμένα από Ωοθήκη Σινικού Κρικητού (Chinese Hamster Ovary).
Η αλληλουχία αμινοξέων της ανασυνδυασμένης μορφής, καθώς και η νουκλεοτιδιακή αλληλουχία κωδικοποίησής της, είναι ταυτόσημες με τη φυσιολογική μορφή της α-γαλακτοσιδάσης A.
Αγαλσιδάση Α (Replagal, Shire)
Αγαλσιδάση Β (Fabrazyme, Genzyme)
#60 Μειωση υποστρωματος στη βιοψια νεφρου σε ασθενη υπο ΕRT
#66 Ιn two clinical trials of enzyme replacement therapy (ERT) with agalsidase alfa, including 37 children, boys demonstrated reductions in plasma Gb3 levels, and both boys and girls reported reductions in neuropathic pain and in the use of neuropathic pain medications. Heart rate variability, which is reduced in boys with Fabry disease, was statistically significantly improved with 6 months of agalsidase alfa treatment. In a single clinical study of agalsidase beta in children (n=16), skin Gb3 deposits and plasma Gb3 levels were reduced in boys. Differences exist in the administration and the safety profile of these two enzyme formulations. Follow-up of these cohorts and additional studies will be necessary to fully evaluate long term efficacy of ERT in children with Fabry disease.
#67 2107467469 Τμήμα Γενετικής στο Χωρέμειο όπου μελετάται το πρότυπο αδρανοποίησης του Χ-χρωμοσώματος σε 3 θέσεις στο χρωμόσωμα που όμως δεν συμπεριλαμβάνουν το γονίδιο της Fabry και η αδρανοποίηση αφορά το αίμα ή τον ιστό (ιστό από παρειά) που θα μελετηθεί. Συνομιλία με κα Δελτρά.
Μεταξύ των τριών χρωμοσωμικών θέσεων που ελέγχονται (3 συνολικά) είναι και η θέση του γονιδίου για τον υποχοχέα ανδρογόνων που έχει διαφορετικό πρότυπο αν είναι πατρικής ή μητρικής προέλευσης το Χ χρωμόσωμα.
#68 Pegunigalsidase alfa has a plasma half-life of about ~ 80 h compared to ~ ≤ 2 h for other currently available ERTs, and carries the theoretical potential for reduced immunogenicity due to epitope masking as suggested by in vitro studies
In female patients and patients with a non-classical disease phenotype, antibody formation against the administered recombinant enzyme is rarely observed
Παράρτημα Fabrazyme: Ανοσογονικότητα Καθώς η αγαλσιδάση βήτα (r-hαGAL) είναι μια ανασυνδυασμένη πρωτεΐνη, αναμένεται ανάπτυξη αντισωμάτων IgG σε ασθενείς με μικρή ή καθόλου υπολειμματική ενζυμική δραστηριότητα. Η πλειονότητα των ασθενών ανέπτυξε αντισώματα IgG στην r-hαGAL, συνήθως εντός 3 μηνών μετά την πρώτη έγχυση Fabrazyme. Με την πάροδο του χρόνου, η πλειονότητα των οροθετικών ασθενών σε κλινικές δοκιμές είτε παρουσίασε μια τάση μείωσης των τίτλων (με βάση μια μείωση του τίτλου κατά ≥ 4 φορές από τη μέγιστη μέτρηση έως την τελευταία μέτρηση) (40% των ασθενών), είτε ανέπτυξε ανοχή [μη ανιχνεύσιμα αντισώματα, πράγμα που επιβεβαιώθηκε με 2 διαδοχικές αναλύσεις ραδιοανοσοκαθίζησης (RIP)] (14% των ασθενών) ή παρουσίασε σταθεροποίηση του τίτλου σε τιμή πλατώ (35% των ασθενών)
The presence of IgG1 and IgG4 anti-r-αGAL A antibodies is associated with in vitro αGAL A activity inhibition.
#72 In patients who have an amenable GLA gene variant, whose baseline eGFR is≥30 mL/min/1.73 m, and who are ≥18 years old (or ≥12 years old if living inthe European Union), both ERT and migalastat are potential options. We engage in shared decision-making regarding the choice between ERT and migalastat.
#84 Μοντελοποίηση GLA μεταλλάξεων μέσω βιοπληροφοριακής ανάλυσης (incilico analysis) όπου ελέγχεται η συγκεκριμένη αλληλούχιση των αμινοξέων της μετάλλαξης και γίνεται πρόβλεψη για τον χαρακτηρισμό της ως παθογόνο κλπ μέσω scors.
Είναι το αντίστοιχο των functional studies όπου η μετάλλαξη προκαλείται σε κυτταροκαλλιέργειες ή πειραματόζωα και ελέγχεται αν παραβλάπτεται η σύνθεση φυσιολογικής πρωτε:ινης.