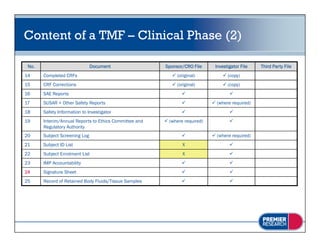
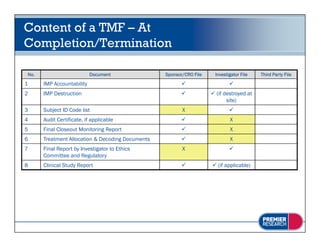
The document discusses the legal and regulatory requirements for managing trial master files, including which documents must be retained, how they should be organized and indexed, and the proper storage conditions and duration of retention. It provides guidance on setting up essential document files for the investigator, sponsor, and third parties. The document also outlines examples of common inspection findings when trial master files are not properly maintained.















![Inspection Findings - Examples
• Inadequate validation [of pivotal computer systems
(e.g.
(e g databases)]
• Lack of source documentation of physical exam,
medical history, concomitant medication, p
y, , primary
y
endpoints, key safety assessments
• Evidence of TMF not being maintained as current
• Sig d consent f
Signed t forms missing
i i g
• Lack of documentation of decisions
• Inadequate facilities for archives and long term
document retention (archivist, security, environmental
control, access control, fire prevention)](https://image.slidesharecdn.com/essentialdocumentsandmanagingtrialfiles-110505012206-phpapp01/85/Essential-documents-and_managing_trial_files-17-320.jpg)














































































