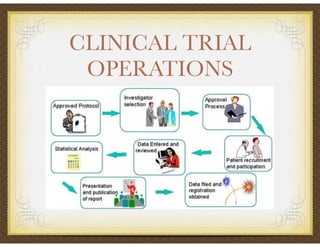
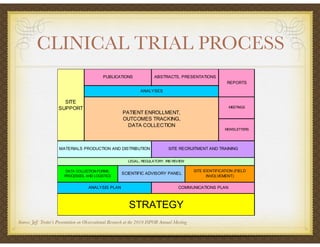
David Selkirk has over 19 years of experience working for large and mid-size pharmaceutical companies and clinical research organizations. He will discuss the landscape of late phase clinical trials, including increasing regulations, the role of health technology assessments, and operational considerations. Key topics that will be covered include working with stakeholders, comparative effectiveness research, risk evaluation and mitigation strategies, patient reported outcomes, and contracting with vendors such as CROs. The future may see larger trials conducted with more independent oversight and accountability.






























































































![R&D Directions Webcast June Final[1]](https://cdn.slidesharecdn.com/ss_thumbnails/rddirectionswebcastjunefinal1-13111948790252-phpapp01-110720160137-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)
![R&D Directions Webcast June Final[1]](https://cdn.slidesharecdn.com/ss_thumbnails/rddirectionswebcastjunefinal1-13111957862724-phpapp02-110720160427-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)