









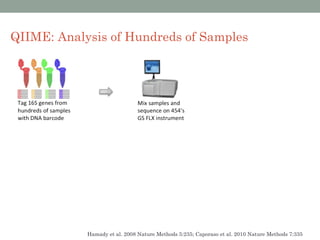


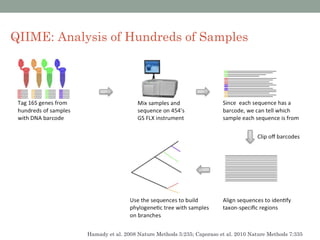
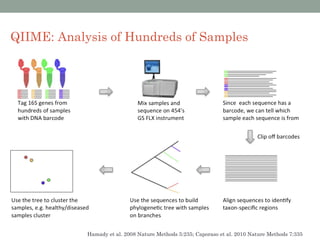

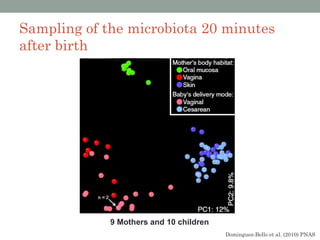
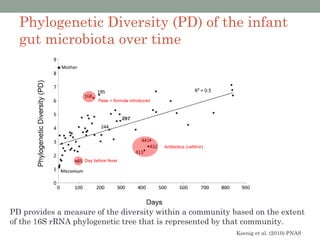
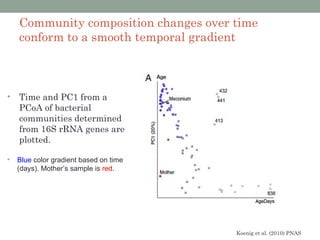
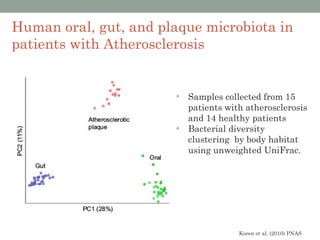
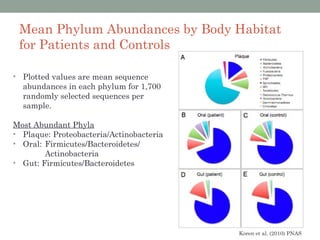
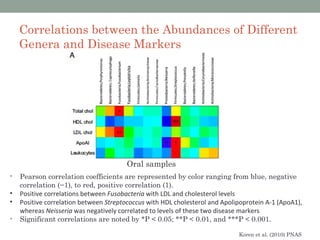

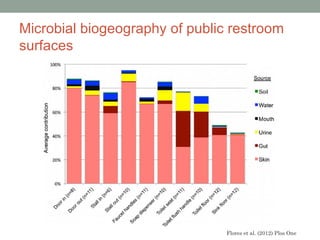


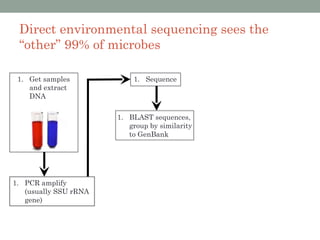
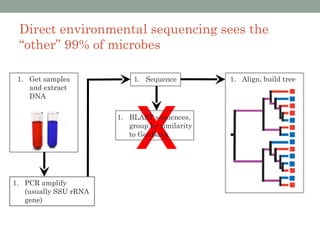
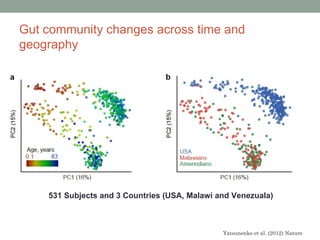
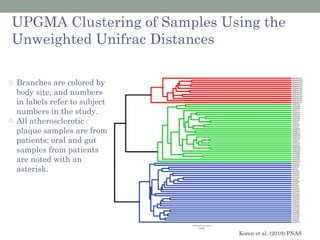
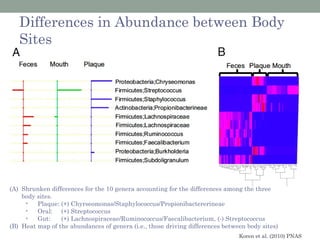

The document discusses high-throughput sequencing techniques for analyzing human microbiome samples. It provides examples of studies that used these techniques to analyze microbiome samples from various body sites and populations. These studies characterized differences in microbial communities between body sites, tracked changes over time, identified correlations with disease markers, and compared communities across geographic locations and diets. Advanced analysis tools like QIIME and UniFrac were highlighted for interpreting vast amounts of sequencing data from hundreds of microbiome samples.



































































































