Downloaded 89 times

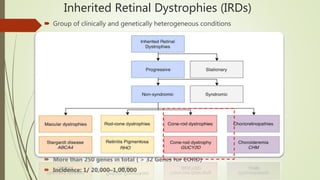
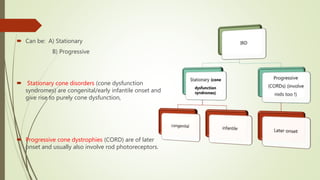

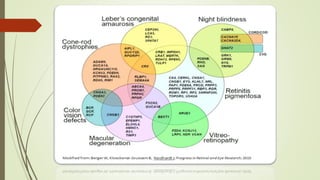



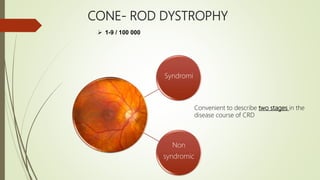






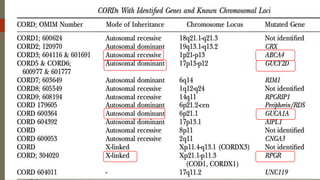


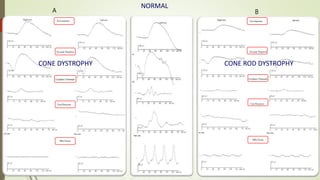




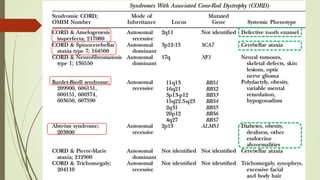

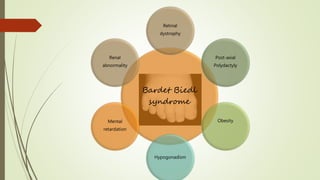








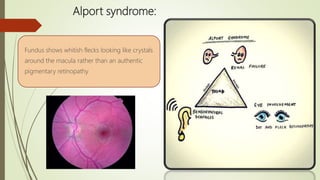
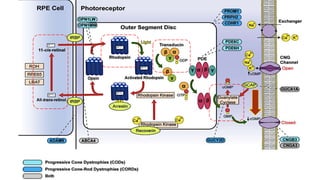
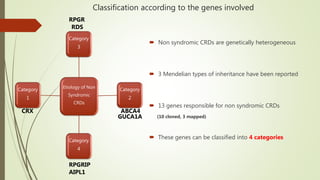






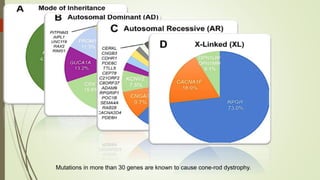
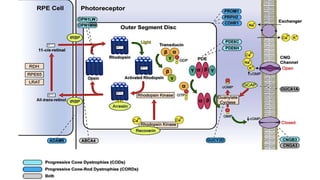















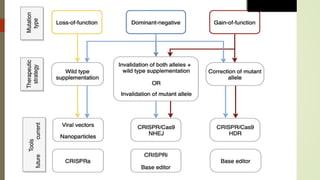




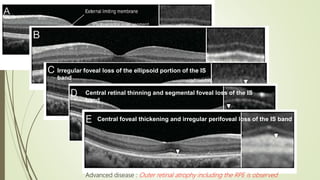
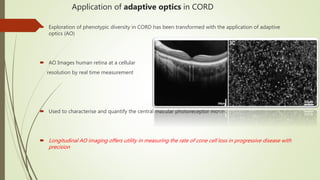


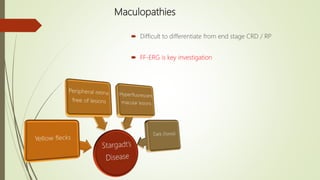


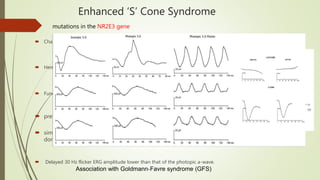

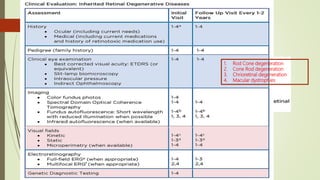



This document provides information about cone and rod dystrophy (CORD), including its genetics, clinical presentation, classification, and mechanisms. It discusses how CORD is a genetically heterogeneous group of inherited retinal dystrophies involving both cone and rod photoreceptors. The document outlines the typical stages and symptoms of CORD and compares it to other conditions like achromatopsia. It also describes syndromic forms of CORD and provides a classification system for CORD based on genetics.























































































