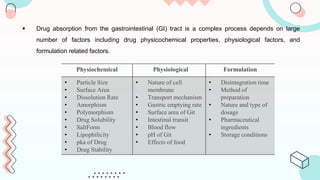
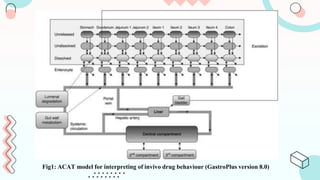
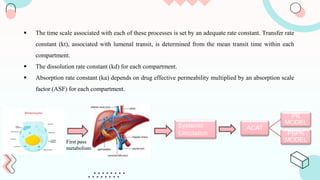
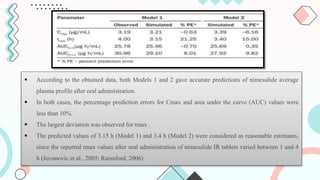
The document discusses computer-aided biopharmaceutical characterization and gastrointestinal absorption simulation, presenting the importance of in silico modeling in drug development. It highlights various models used for simulating gastrointestinal absorption, including the advanced dissolution, absorption, and metabolism (ADAM) model and the gastrointestinal transit-absorption (GITA) model, emphasizing their role in predicting pharmacokinetics. Additionally, the document addresses the impact of fasting and feeding states on drug absorption, the significance of in vitro-in vivo correlation (IVIVC), and the considerations for biowaivers in drug approval processes.