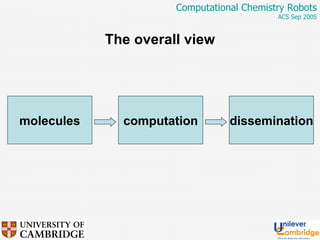
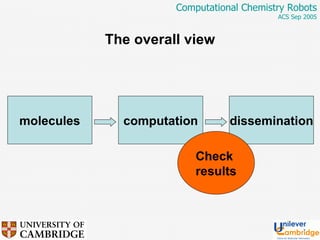
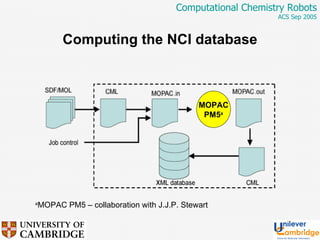
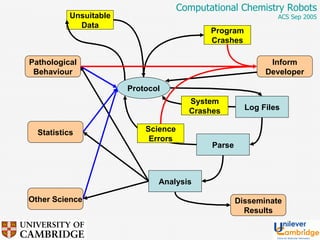
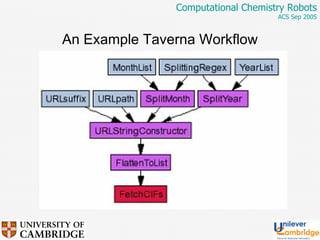
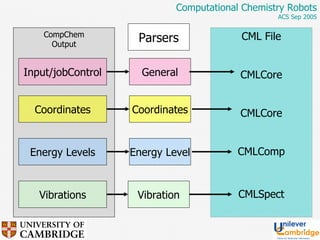
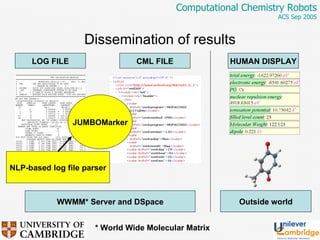
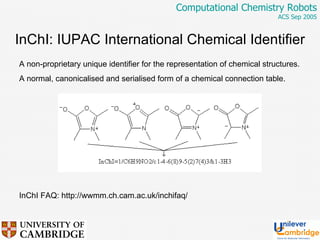
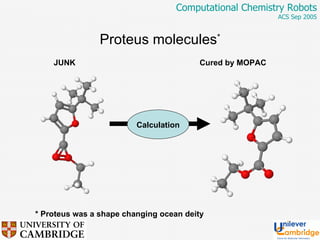
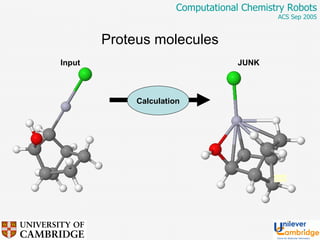
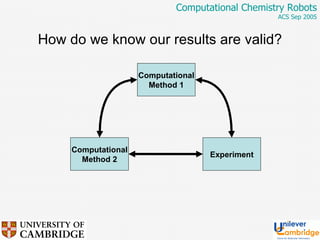
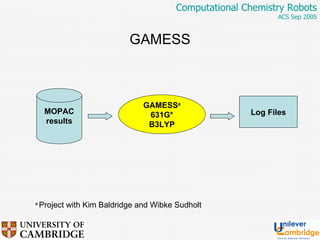
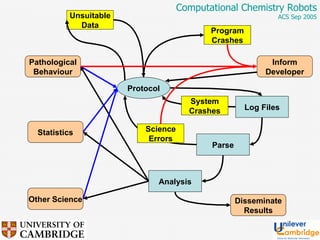
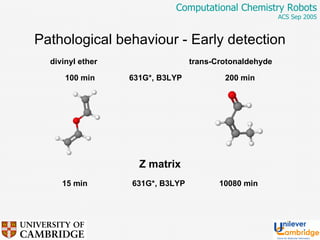
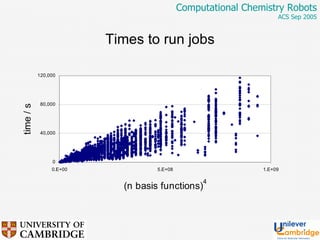
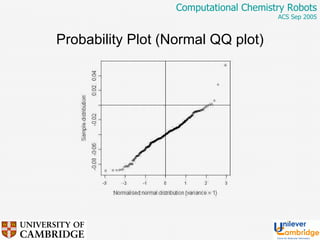
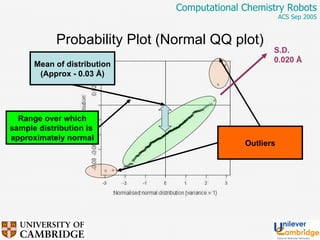
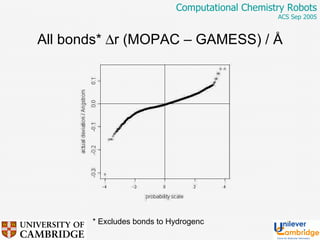
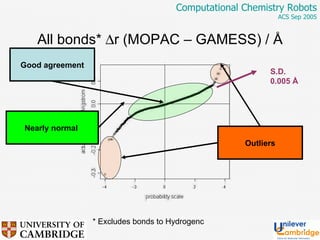
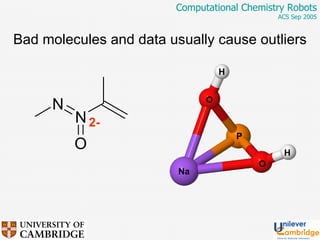
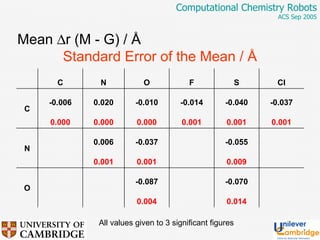
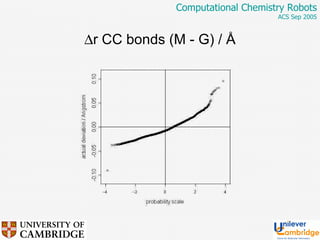
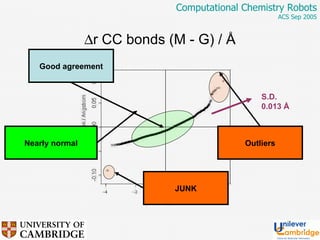
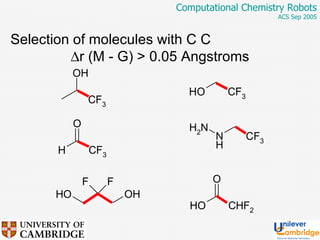
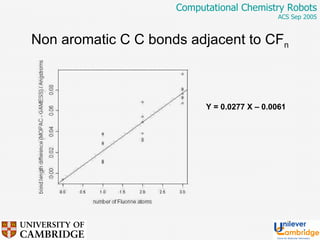
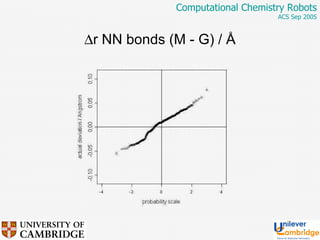
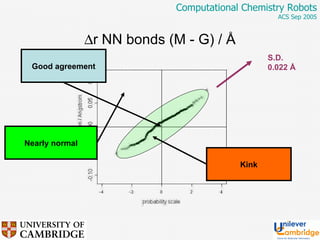
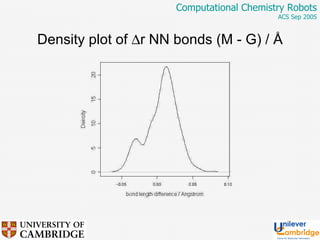
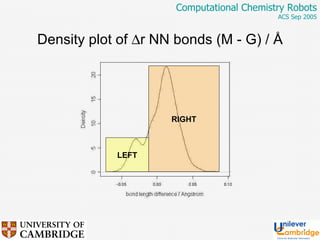
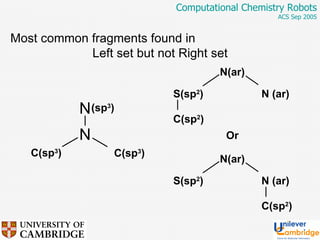
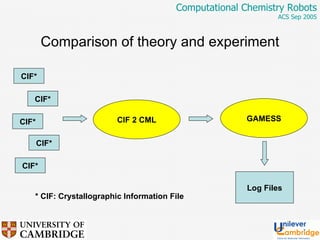
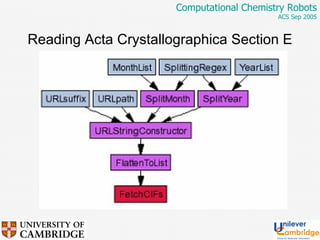
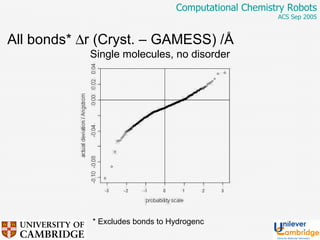
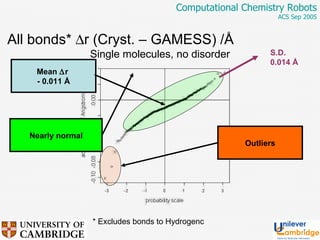
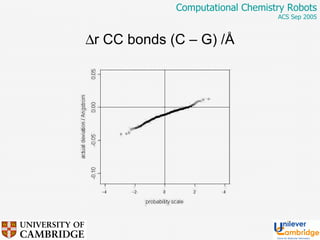
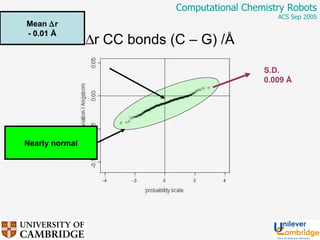
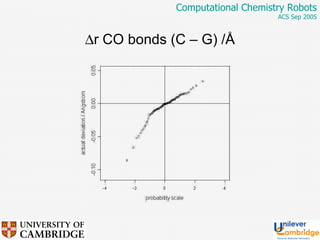
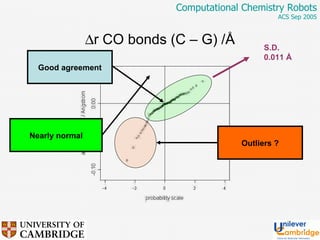
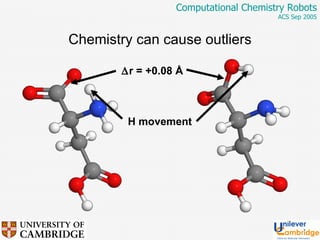
This document discusses the automation of computational chemistry calculations and protocols to reliably generate molecular property data. It addresses validating computational methods, analyzing results for errors and outliers, and comparing output to experimental data. The goal is to provide high-quality "experimental" data through automated high-throughput computation while ensuring valid results and identifying unusual computations. Workflows, parsing tools, and dissemination methods are presented for managing large numbers of jobs and analyzing results.
![Computational Chemistry Robots ACS Sep 2005 Computational Chemistry Robots J. A. Townsend, P. Murray-Rust, S. M. Tyrrell, Y. Zhang [email_address]](https://image.slidesharecdn.com/acs2005final-091211041113-phpapp02/85/Computational-Chemistry-Robots-1-320.jpg)