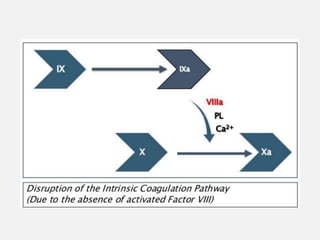
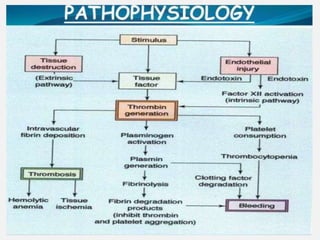
Hereditary and acquired coagulation disorders are caused by deficiencies in clotting factors that lead to defects in normal clot formation. Hereditary disorders include hemophilia A/classical hemophilia due to factor VIII deficiency and hemophilia B/Christmas disease due to factor IX deficiency. Von Willebrand's disease is the most common hereditary disorder characterized by qualitative or quantitative defects in von Willebrand factor. Acquired disorders involve deficiencies of multiple clotting factors and can be caused by vitamin K deficiency, liver disease, or disseminated intravascular coagulation. Coagulation disorders are diagnosed through prolonged coagulation tests and assays showing reduced clotting factor levels. Treatment focuses on replacing the