LEARNING OBJECTIVES
• Definecongenital abnormalities
• List types of congenital abnormalities of the
GIT
Causes of congenital abnormalities
Signs and symptoms of each congenital
abnormalities
Management of each congenital abnormalities
Complications of congenital abnormalities
3.
Definition
• Congenital anomaliescan be defined as
structural or functional anomalies that occur
during intrauterine life. Also called birth
defects, congenital disorders, or congenital
malformations, these conditions develop
prenatally and may be identified before or at
birth, or later in life
4.
incidence
• An estimated6% of babies worldwide are
born with a congenital anomaly, resulting in
hundreds of thousands of associated deaths.
However, the true number of cases may be
much higher because statistics do not often
consider terminated pregnancies and
stillbirths.
5.
• Some congenitalanomalies can be treated
with surgical and non-surgical options, such as
cleft lip and palate, clubfoot, and hernias.
Others, including heart defects, neural tube
defects, and down syndrome, can cause
lifelong impacts.
6.
Causes of congenitalabnormalities
• Approximately 50% of congenital anomalies
cannot be linked to a specific cause. However,
known causes include
• single gene defects, chromosomal disorders,
• multifactorial inheritance
• environmental teratogens
• micronutrient deficiencies
• Genetic causes can be traced to inherited genes
7.
The vast majority(94%) of congenital anomalies
occur in low- and middle-income countries. Possible
causes include
• lack of screening and prenatal care, insufficient
access to nutritious foods for pregnant women
• use or contact with alcohol or tobacco
• increased exposure to infection
• environmental contaminants
8.
Causes cont…..
Some drugsmay cause cleft lip and cleft palate.
Among them: anti-seizure/anticonvulsant
drugs, acne drugs containing Accutane, and
methotrexate, a drug commonly used for
treating cancer, arthritis, and psoriasis
9.
cont
• Consanguinity –when parents are related by
blood – increases the risk of congenital
anomalies and nearly doubles the risk of
neonatal and early childhood death,
intellectual disability and other health
conditions.
• Advanced maternal age also increases the risk
of chromosomal abnormalities
10.
cont
including Down syndrome.
Somediseases are known to cause increases in
rates of congenital anomalies including Zika
virus, syphilis and rubella. Other anomalies,
such as cystic fibrosis and haemophilia C, are
more prevalent in specific ethnic
communities.
11.
PREVENTION OF CONGNITAL
ABNORMALITIES
•Some congenital anomalies can be prevented
through;
• screening
• vaccination
• fortification of staple foods with nutrients such
as folic acid and iodine
• adequate antenatal care, among other methods.
• Having enough rest and sleep
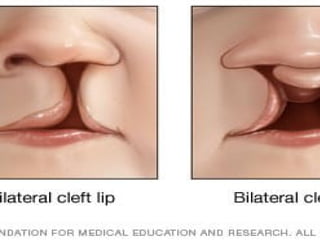
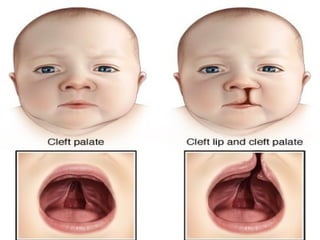
Cleft palate andhare or cleft lip.
Defination
Cleft lip and cleft palate are openings or splits in
the upper lip, the roof of the mouth (palate) or
both. Cleft lip and cleft palate result when facial
structures that are developing in an unborn
baby don't close completely.
16.
Cont…..
• Cleft lipand cleft palate are among the most
common birth defects. They most commonly
occur as isolated birth defects but are also
associated with many inherited genetic
conditions or syndromes.
17.
Cont…
• Having ababy born with a cleft can be
upsetting, but cleft lip and cleft palate can be
corrected. In most babies, a series of surgeries
can restore normal function and achieve a
more normal appearance with minimal
scarring.
18.
Signs and symptoms.
•A split in the lip and roof of the mouth (palate)
that affects one or both sides of the face
• A split in the lip that appears as only a small
notch in the lip or extends from the lip
through the upper gum and palate into the
bottom of the nose
• A split in the roof of the mouth that doesn't
affect the appearance of the face
19.
Signs & symptomscontinuation
• Difficulty with feedings
• Difficulty swallowing, with potential for liquids
or foods to come out the nose
• Nasal speaking voice
• Chronic ear infections
20.
ASSESSMENT AND DIAGNOSTICFINDING
The physical appearance of the newborn confirms the
diagnosis of cleft lip; diagnosis of cleft palate is made at
birth.
• Inspection.
Diagnosis of cleft palate is made at birth with the
close inspection of the newborn’s palate; to be
certain that a cleft palate is not missed, the
examiner must insert a gloved finger into the
newborn’s mouth to feel the palate to determine that it is
intact.
• Observation.
Cleft lip can be diagnosed through observation of the
physical appearance of the newborn.
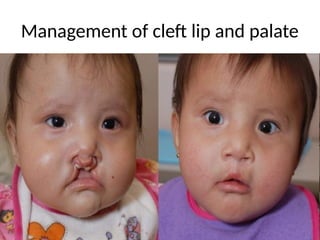
Management of cleftpalate and lip
Aims of management
• To improve the child's ability to Feed, speak
and hear
• To achieve a normal facial appearance
23.
Management
• Treatment involvessurgery to repair the defect
and therapies to improve any related conditions.
• cleft lip repair — within the first 3 to 6 months
of age
• Cleft palate repair — by the age of 12 months,
or earlier if possible
• Follow-up surgeries — between age 2 and late
teen years
24.
Cleft palate repair
•Various procedures may be used to close the
separation and rebuild the roof of the mouth
depending on your child's situation.
• The surgeon makes incisions on both sides of
the cleft and repositions the tissue and
muscles. The repair is then stitched closed.
25.
Cleft lip repair
•To close the separation in the lip, the surgeon
makes incisions on both sides of the cleft and
creates flaps of tissue.
• The flaps are then stitched together, including
the lip muscles.
• The repair should create a more normal lip
appearance, structure and function.
• Initial nasal repair, if needed, is usually done at
the same time.
26.
Ear tube surgery
•For children with cleft palate, ear tubes may
be placed to reduce the risk of chronic ear
fluid, which can lead to hearing loss.
• Ear tube surgery involves placing tiny bobbin-
shaped tubes in the eardrum to create an
opening to prevent fluid buildup.
27.
Surgery to reconstructappearance
• Additional surgeries may be needed to
improve the appearance of the mouth, lip and
nose.
• To improve your child's appearance, quality of
life, and ability to eat, breathe and talk.
28.
Nursing care
• Aims
•Maintaining adequate nutrition.
• Increasing family coping.
• Reducing the parents’ anxiety and guilt
regarding the newborn’s physical defects, and
preparing parents for the future repair of the
cleft lip and palate.
29.
Nursing interventions
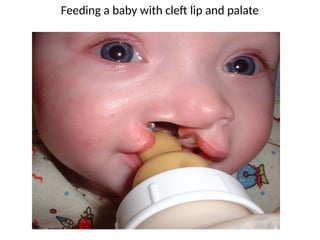
• Maintainadequate nutrition.
Breastfeeding may be successful because the
breast tissue may mold to close the gap; if the
newborn cannot be breastfeed, the mother’s
breast milk may be expressed and used instead
of formula; a soft nipple with a cross-cut made
to promote easy flow of milk may work well.
30.
Interventions cont..
• Positioning.
Ifthe cleft lip is unilateral, the nipple should be
aimed at the unaffected side; the infant should be
kept in an upright position during feeding.
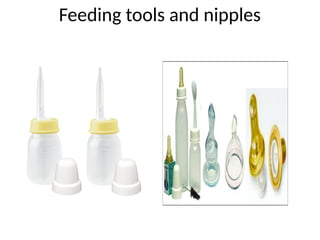
• Tools for feeding. Lamb’s nipples
(extra long nipples) and special cleft palate nipples
molded to fit into the open palate area to close the gap
may be used; one of the simplest and most effective
methods may be the use of an eyedropper or an Asepto
syringe with a short piece of rubber tubing on the tip
INTERVENTIONS CONT..
• Promotefamily coping.
Encourage the family to verbalize their feelings
regarding the defect and their disappointment;
serve as a model for the family caregiver’s
attitudes toward the child.
34.
Interventions cont…
• Reducefamily anxiety.
Give the family information about cleft repairs;
encourage them to ask questions and reassure
them that any question is valid.
• Provide family teaching.
Explain the usual routine of preoperative,
Intra operative, and post operative care; written
information is helpful, but be certain the parents
understand the information.
35.
Prevention of cleftlip and palate
Take folic acid before pregnancy and during
early pregnancy ie 1st
tremister
This help to protect the baby from cleft lip and
palate and other birth defects of the brain and
spine called neural tube defects.
Learning objectives
• Defineesophageal atresia
• Types of oesophageal atresia
• Cause of oesophageal atresia
• Signs and symptoms of oesophageal atresia
• Management of oesophageal atresia
39.
Esophageal atresia.
Definition
• abirth defect in which part of a baby's
esophagus does not develop properly, the
is the tube that connects the mouth to the
stomach
• Esophageal atresia is a birth defect of the
swallowing tube (esophagus) that connects
the mouth to the stomach.
40.
Cont…
• Instead offorming a tube between the mouth
and the stomach, the esophagus grows in two
separate segments that do not connect. In
some children, so much of the esophagus is
missing that the ends can't be easily
connected with surgery. This is known as long-
gap .
41.
High risk ofhaving babies esophageal atresia
• Fathers who are older at the time of the
baby’s conception.
• Women who have undergone fertility
treatments, including intrauterine
insemination and in vitro fertilization.
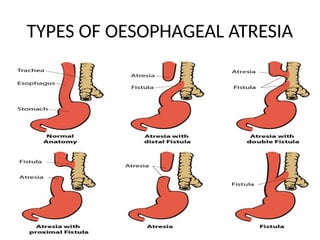
Types of oesophagealatresia
Four types of esophageal atresia
• Type A
is when the upper and lower parts of the
esophagus do not connect and have closed ends. In
this type, no parts of the esophagus attach to the
trachea.
• Type B
is very rare. In this type the upper part of the
esophagus is attached to the trachea, but the lower
part of the esophagus has a closed end.
44.
Continuation
• Type C
themost common type. In this type the upper
part of the esophagus has a closed end and the
lower part of the esophagus is attached to the
trachea, as is shown in the drawing.
• Type D
the rarest and most severe. In this type the upper
and lower parts of the esophagus are not
connected to each other, but each is connected
separately to the trachea.
45.
Causes of oesophagealatresia
• The causes of esophageal atresia in most babies are
unknown.
• Researchers believe that some instances of
esophageal atresia may be caused by abnormalities
in the baby’s genes.
• Nearly half of all babies born with esophageal atresia
have one or more additional birth defects, such as
other problems with the digestive system (intestines
and anus), heart, kidneys, or the ribs or spinal
46.
continuation
• Paternal age– Older age of the father is
related to an increased chance of having a
baby born with esophageal atresia.
• Women who used ART to become pregnant
have an increased risk of having a baby with
esophageal atresia compared to women who
did not use ART.(assisted reproductive
technology)
47.
Signs and symptoms
•frothy white bubbles in your baby's mouth
• coughing or choking when feeding
• blue color of the skin, especially when your
baby is feeding
• difficulty breathing
48.
Diagnosis
• Esophageal atresiais most commonly
detected after birth when the baby first tries
to feed and has choking or vomiting, or when
a tube inserted in the baby’s nose or mouth
cannot pass down into the stomach.
• An x-ray can confirm that the tube stops in
the upper esophagus.
49.
Management of esophageal
•Esophageal atresia can be life-threatening, so
the baby has to be treated quickly.
• Doctors perform surgery to connect the
esophagus to the stomach in babies with this
condition
50.
Mgt cont…
• Babieswho are otherwise healthy have
surgery just a few days after they are born.
• Babies with other health issues or disabilities
at birth may need to wait to have surgery for
esophageal atresia. If your baby has to wait for
surgery, they will receive nutrition through an
IV (a tiny tube inserted into a vein) until the
operation occurs.
51.
Mgt cont…..
• Oncea diagnosis has been made, surgery is
needed to reconnect the two ends of the
esophagus so that the baby can breathe and
feed properly.
• Multiple surgeries and other procedures or
medications may be needed, particularly if the
baby’s repaired esophagus becomes too
narrow for food to pass through it;
52.
Complications of esophagealatresia
• About half of all babies with esophageal
atresia also have other congenital disabilities
such as heart, kidney and spinal problems.
53.
Complications after surgery
•Gastroesophageal reflux disease (GERD): Acid
from the stomach travels back up into the
esophagus, which can lead to inflammation and
a burning sensation.
• Scar tissue: Scar tissue can form in the area
where the esophagus is surgically repaired,
leading to narrowing and swallowing difficulty.
• Tracheomalacia: Windpipe walls are weak and
floppy, causing noisy, high-pitched breathing.
54.
Prevention of esophagealatresia
Taking folic acid before and after conception
Eating healthy foods.
• Exercising.
• Getting enough rest.
• Seeing your provider regularly.
ANAL ATRESIA
Learning objectives
•Define anal atresia
• Types of anal atresia
• Causes of anal atresia
• Signs and symptoms of anal atresia
• Management of anal atresia
57.
Definition of analatresia
• is a congenital anorectal malformation (ARM)
where a normal anal opening is absent at birth.
ARMs comprise of a broad spectrum of defects
ranging from minor (e.g., membranous covering)
to complex cloacal malformations involving the
urinary and genital tracts as well.8 Sept 2022 or
• refers to a spectrum of anorectal abnormalities
ranging from a membranous separation to
complete absence of the anus.
58.
Clinical presentation
• Clinicallythere is no anal opening and failure to pass
meconium.
• Abdominal distension
• Sever abdominal pain
• Difficult inbreathing
59.
Types of atresia
•Malformations found in both males and females
• imperforate anus without fistula – the anal
opening is missing or in the wrong place
• rectal atresia and stenosis –the anus or rectum is
too small to allow stool to pass
• rectoperineal fistula –the rectum connects to the
perineum, an area of skin between the anus and
genitals
60.
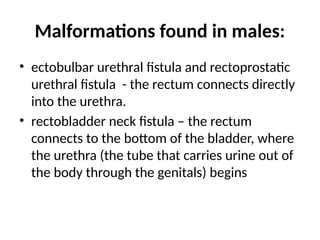
Malformations found inmales:
• ectobulbar urethral fistula and rectoprostatic
urethral fistula - the rectum connects directly
into the urethra.
• rectobladder neck fistula – the rectum
connects to the bottom of the bladder, where
the urethra (the tube that carries urine out of
the body through the genitals) begins
61.
Malformations found infemales:
• recto vestibular fistula - the rectum connects
to just outside of the vagina
• cloaca –the vagina, rectum and urinary
tractare combined into a single channel
• Causes
• Un known
• Chromosomal abnormalities.
62.
Diagnosis of analatresia
• Abdominal radiograph
• can be variable depending on the site of atresia (i.e.
high or low), level of meconium impaction and
physiological effects such as straining
• may show multiple dilated bowel loops with an
absence of rectal gas
• air within urinary bladder suggests high type 6
• calcified meconium in the bowel loops would suggest
high type (meconium calcifies due to urine exposure) 6
63.
Dx cont……
• Ultrasound
•the anus may be seen as an echogenic spot at the
level of the perineum and in anal atresia, this
echogenic spot may be absent 4
• may show bowel dilatation
• an infracoccygeal or transperineal approach may
allow differentiation between high and low subtypes 4
64.
Dx cont…..
MRI
• Canbe used pre/post-operatively to study
pelvic floor, renal, and spinal abnormalities
65.
Treatment and prognosis
•low subtypes are treated with anoplasty
(Surgery to repair or reconstruct the anus)
• high subtypes are treated with colostomy with
subsequent potential repair
PYLORIC STENOSIS
Learning objectives
•Definition of pyloric stenosis
• Causes of pyloric stenosis
• Signs and symptoms of pyloric stenosis
• Management of pyloric stenosis
68.
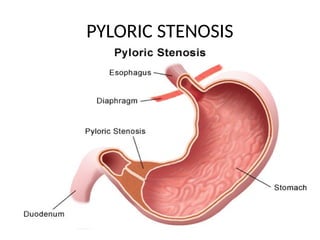
Definition
• Pyloric stenosisis an uncommon condition in
infants that blocks food from entering the small
intestine.
• Normally, a muscular valve (pylorus) between the
stomach and small intestine holds food in the
stomach until it is ready for the next stage in the
digestive process. In pyloric stenosis, the pylorus
muscles thicken and become abnormally large,
blocking food from reaching the small intestine.
Causes of pyloricstenosis
• The causes of pyloric stenosis are unknown
• genetic and environmental factors might play
a role. Pyloric stenosis usually isn't present at
birth and probably develops after ward.
71.
Risk factors.
• Sex.Pyloric stenosis is seen more often in boys — especially
firstborn children — than in girls.
• Race. Pyloric stenosis is more common in whites of northern
European ancestry, less common in Black people and rare in Asians.
• Premature birth. Pyloric stenosis is more common in babies born
prematurely than in full-term babies.
• Family history. Studies found higher rates of this disorder among
certain families. Pyloric stenosis develops in about 20% of male
descendants and 10% of female descendants of mothers who had
the condition.
• Smoking during pregnancy. This behavior can nearly double the
risk of pyloric stenosis.
72.
Continuation of risks
•Pyloric stenosis is a problem that affects
babies between birth and 6 months of age and
causes forceful vomiting that can lead to
dehydration. It is the second most common
problem requiring surgery in newborns.
73.
continuations
• The lowerportion of the stomach that
connects to the small intestine is known as the
pylorus. In pyloric stenosis, the muscles in this
part of the stomach enlarge, narrowing the
opening of the pylorus and eventually
preventing food from moving from the
stomach to the intestine.
74.
Continuation of riskfactors.
•
Early antibiotic use. Babies given certain
antibiotics in the first weeks of life —
erythromycin to treat whooping cough, for
example — have an increased risk of pyloric
stenosis.
• Bottle-feeding. Some studies suggest that
bottle-feeding rather than breast-feeding can
increase the risk of pyloric stenosis.
INVESTIGATIONS
• Blood teststo check for dehydration or
electrolyte imbalance or both.
• Ultrasound to view the pylorus and confirm a
diagnosis of pyloric stenosis.
• X-rays of your baby's digestive system, if
results of the ultrasound aren't clear.
77.
After surgery
• Thebaby is given intravenous fluids for a few
hours. The baby can start feeding again within
12 to 24 hours.
• The baby might want to feed more often.
• Some vomiting may continue for a few days.
78.
SURGERY
Surgery is neededto treat pyloric stenosis.
pyloromyotomy, the surgeon cuts only
through the outside layer of the thickened
pylorus muscle, allowing the inner lining to
bulge out. This opens a channel for food to
pass through to the small intestine.
79.
Complications
• Failure togrow and develop.
• Dehydration. Frequent vomiting can cause
dehydration and a mineral (electrolyte)
imbalance
• Stomach irritation. Repeated vomiting can
irritate the baby's stomach and may cause mild
bleeding.
• Jaundice. liver (bilirubin) can build up, causing a
yellowish discoloration of the skin and eyes.
Imperforate anus.
• Learningobjectives.
• Definition of imperforate anus.
• Types of imperforate anus.
• Causes of imperforate anus
• Risk factors of imperforate anus.
• Signs and symptoms.
• Management of imperforate anus.
• Complication.
82.
Definition of imperforateanus
• An imperforate anus happens when the anus is
missing or doesn't have a hole.
• The anus is the muscle ring that lets a person
hold poop inside, then release it later during a
bowel movement.
• Imperforate anus is a type of birth defect called
an anal malformation. This means that the
anus and rectum don't form in the usual way.
83.
TYPES OF IMPERFORATEDANUS
Malformations found in both males and
females:
• Anorectal malformation without fistula – the
anal opening is missing or in the wrong place
• Rectal atresia and stenosis – the anus or rectum
is too small to allow stool to pass
• Rectoperineal fistula – the rectum connects to
the perineum, an area of skin between the anus
and genitals
84.
MALFORMATIONS FOUND INMALES
• Rectobulbar urethral fistula and rectoprostatic
urethral fistula – the rectum connects directly
into the urethra (the tube that carries urine out
of the body through the genitals)
• Recto-bladder neck fistula – the rectum
connects to the bottom of the bladder, where
the urethra begins
85.
MALFORMATIONS FOUND INFEMALES
• Rectovestibular fistula – the rectum connects
to just outside of the vagina
• Rectovaginal fistula – rare malformation with
a connection between the rectum and the
vagina
• Cloaca – the vagina, rectum and urinary tract
are combined into a single channel
86.
Causes of imperforateanus.
• Imperforate anus is a birth defect that usually
appears to occur randomly for unknown
reasons (sporadically). Less commonly, the
condition may be familial, suggesting
autosomal dominant, autosomal recessive, or
X-linked recessive inheritance
87.
Risk factors ofimperforate anus.
• Many genes may play a role in causing
imperforate anus.
• Environmental factors may also play a role in
this condition. These include exposure to
alcohol, substances
• smoking
• some infections.
88.
Signs and symptomsof imperforate
anus
• The opening to the anus is missing or not in
the usual place. In girls, it may be near the
vagina.
• No passage of poop within a day or 2 of birth.
• Passing poop through another opening, like
the urethra in boys or vagina in girls.
• Swollen belly.
89.
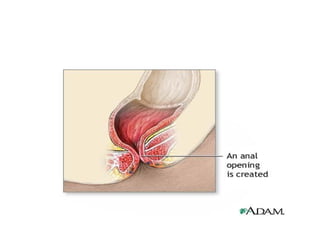
Management
• Imperforate anusrepair
anus repair is surgery to correct a birth defect
involving the rectum and anus.
• An imperforate anus defect prevents most or
all stool from passing out of the rectum.
90.
• How thissurgery is performed depends on the
type of imperforate anus.
• The surgery is done under general anesthesia.
This means the infant is asleep and feels no
pain during the procedure.
92.
For mild imperforateanus defects:
• The first step involves enlarging the opening where
the stool drains, so stool can pass more easily.
• Surgery involves closing any small tube-like openings
(fistulas), creating an anal opening, and putting the
rectal pouch into the anal opening. This is called an
anoplasty.
• The child must often take stool softeners for weeks
to months.
93.
• Two surgeriesare often needed for more severe
imperforate anus defects:
• The first surgery is called a colostomy. The
surgeon creates an opening (stoma) in the skin
and muscle of the abdominal wall. The end of the
large intestine is attached to the opening. Stool
will drain into a bag attached to the abdomen.
• The baby is often allowed to grow for 3 to 6
months.
94.
Continuation of management
•In the second surgery, the surgeon moves the
colon to a new position. A cut is made in the
anal area to pull the rectal pouch down into
place and create an anal opening.
• The colostomy will likely be left in place for 2
to 3 more months.
• child's surgeon can tell more about the exact
way the surgeries will be done.
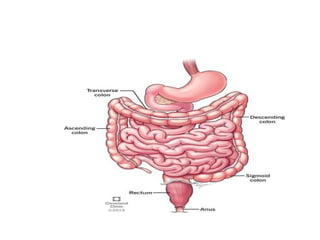
Definition
Abnormally large ordilated colon due to congenital
absence of myenteric ganglion cells in a distal
segment of the large bowel.
OR
Congenital MEGACOLON resulting from the absence of
ganglion cells (aganglionosis) in a distal segment of the
LARGE INTESTINE. The aganglionic segment is
permanently contracted thus causing dilatation proximal
to it. In most cases, the aganglionic segment is within the
RECTUM and SIGMOID COLON.
99.
What causes megacolon?
• Mega colon has a wide range of causes,
however, the condition is often idiopathic,
which means the exact cause is not known.
• Other causes including infection, disease,
medication, and various congenital disorders.
It may also occur following a major surgery
100.
Continuation of causes
•Infection
One of the most common causes of megacolon
is infection. This includes bacterial infections
such as Clostridium difficile, Salmonella,
Shigella, and Campylobacter, as well as parasitic
infections such as Trypanosoma cruzi
(commonly known as Chagas disease) and
Entamoeba histolytica.
101.
continuation
• Disease
Mega coloncan also be caused by a variety of
systemic diseases. These include some muscular
dystrophies,
scleroderma, and systemic lupus
erythematosus.
102.
Continuation of causes
Medication
Inrare cases, mega colon may be the adverse
effect of a medication. Most notably, drugs such
as clozapine, and loperamide are
associated with increased risk of mega colon.
103.
Continuation of causes
Congenitaldisorders
Mega colon can also be caused by some
congenital disorders, as is true in the case of
Hirschsprung’s disease, where functional
obstruction of the intestines is often observed.
Signs and symptomsof mega colon
Constipation
bloating
Abdominal pain or tenderness.
In more severe cases, hard fecal masses called
fecalomas may also be present.
106.
Continuation of signsand symptom
• Depending on the cause, mega colon may
have additional symptoms. In toxic mega
colon, usually caused by infection, additional
symptoms include fever, tachycardia, and
shock. In disease-related cases of mega colon,
additional symptoms are those of the disease
itself.
107.
TYPES OF MEGACOLON
Can be classified as acute or chronic depending
on whether the dilation is temporary or
ongoing. All cases of acute mega colon are
acquired, whereas chronic mega colon can be
both acquired or congenital.
108.
Management of megacolon
• Treatment for mega colon starts by addressing
the underlying cause (such as the offending
medication or disease), if known.
In acute mega colon, all food and drink should be
withheld and a nasogastric tube placed. If non-
toxic, neostigmine should be administered, and if
necessary, the colon itself should be decompressed
by means of a colonoscopy. If toxic, steroids and
broad spectrum antibiotics should be given.
109.
Cont… of management
•Bowel rest and bowel decompression. These
treatments remove gas and substances filling
the colon.
• IV fluids. may be given, an IV of fluids and
electrolytes to help nourish your body and
prevent dehydration.
• Surgery.
110.
Mgt cont…
• Inchronic mega colon, both dietary
and pharmacological methods
should be used to increase intestinal
motility.
• Laxatives and enemas may also be
used to prevent fecal impaction.
111.
Mgt cont…
• Ifthe patient does not respond to these
treatments within one to three days, it may be
necessary to use surgery to remove all or part
of the colon.
• Following colectomy, options include ileorectal
anastomosis and ileostomy.
Constipation acute orchronic.
• Learning objectives
• Definition of constipation
• Types of constipation
• Risk factors of constipation.
• Causes of constipation
• Signs and symptoms
• Management of constipation.
114.
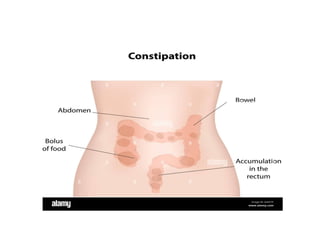
Definition
• Is whenbowel movements become less frequent
and stools become difficult to pass.
• It happens most often due to changes in diet or
routine, or due to inadequate intake of fiber.
• in severe pain, blood in stools, or constipation
that lasts longer than three weeks visit the
hospital
• Acute last few days.
• Chronic last for several weeks or months
117.
Types of constipation
•Normal transit constipation
Normal transit constipation is a condition in
which a person perceives themselves to be
constipated, but the consistency of their stools
is normal, and the stools move through the
digestive tract at a regular pace.
118.
Types cont..
• Slowtransit constipation
• People with slow transit constipation do not
experience the normal stimulation of the
bowels, called peristalsis, after eating.
Therefore, food moves through the digestive
tract more slowly than usual, and stools take
longer to pass through the colon.
119.
Types cont..
• Outletconstipation
occurs as a result of damage to the pelvic floor
muscles. These muscles support the bowel and
bladder.
120.
Types cont..
• Secondaryconstipation
Secondary constipation is constipation
That occurs as a result of an underlying health
issue or a side effect of medication use. The
most common causes of secondary
constipation include;
• diseases that affect the brain or blood vessels
• the use of certain medications
121.
Risk factors.
• sedentary
•not eating enough fiber
• not drinking enough fluids
• medications
• Have a medical condition affecting the anus or
rectum
• Have a neurological disorder
122.
Risks cont..
• Natalsex: Females are more likely than males
to develop constipation.
Use of laxatives and enemas: Prolonged use of
these constipation treatments may make it
more difficult to have a bowel movement
without them.
123.
Causes
• Medications. drugscan contribute to constipation.
• Cow's milk allergy. An allergy to cow's milk or consuming
too many dairy products (cheese and cow's milk) sometimes
leads to constipation.
• Family history. Children who have family members who
have experienced constipation are more likely to develop
constipation. This may be due to shared genetic or
environmental factors.
• Medical conditions. Rarely, constipation in children
indicates an anatomic malformation, a metabolic or
digestive system problem, or another underlying condition.
124.
continuation
• Changes inroutine. Any changes in the child's
routine — such as travel, hot weather or stress —
can affect bowel function. Children are also more
likely to experience constipation when they first
start school outside of the home. Withholding. The
child may ignore the urge to have a bowel
movement because he or she is afraid of the toilet
or doesn't want to take a break from play. Some
children withhold when they're away from home
because they're uncomfortable using public toilets.
125.
continuation
• Painful bowelmovements caused by large, hard stools also may
lead to withholding. If it hurts to pass stool, the child may try to
avoid a repeat of the distressing experience.
• Toilet training issues. If you begin toilet training too soon, the child
may rebel and hold in stool. If toilet training becomes a battle of
wills, a voluntary decision to ignore the urge to pass stool can
quickly become an involuntary habit that's tough to change.
• Changes in diet. Not enough fiber-rich fruits and vegetables or fluid
in the child's diet may cause constipation. One of the more
common times for children to become constipated is when they're
switching from an all-liquid diet to one that includes solid foods.
126.
Signs and symptoms
•Less than three bowel movements a week
• Bowel movements that are hard, dry and difficult
to pass
• Pain while having a bowel movement
• Stomach pain
• Traces of liquid or pasty stool in the child's
underwear — a sign that stool is backed up in the
rectum
• Blood on the surface of hard stool
128.
Management
The most effectivetreatment will depend on the
type of constipation that is normal or slow
transit constipation or outlet constipation.
129.
Mgt cont..
• Normaland slow transit constipation
often respond well to changes to everyday routines,
such as:
• increasing fiber intake by eating more fruits,
vegetables, and whole grains
• drinking more water
• getting more exercise
• In some cases, laxatives are recommended. These
work to increase bowel movements or loosen stools.
130.
Outlet constipation
• biofeedbacktherapy.
• In biofeedback therapy, a trained therapist
inserts a probe into the anal sphincter. The
therapist then gives visual or verbal feedback
about how the person is using their pelvic floor
muscles and anal sphincter during bowel
movements. This information helps the person
retrain the pelvic floor muscles to improve
their coordination.
131.
• The treatmentfor secondary constipation
begins with identifying and treating the cause.
132.
Also changes mayhelp;
• increasing physical activity
• eating more fiber
• drinking more fluids
• In some cases, a person with secondary
constipation may need surgery to repair or
remove a dysfunctional part of their colon.
133.
complications
• Painful breaksin the skin around the anus
(anal fissures)
• Rectal prolapse, when the rectum comes out
of the anus
• Stool withholding
134.
Complication cont..
• haemorrhoids(piles)
• faecal impaction (where dry, hard stools
collect in the rectum)
• bowel incontinence (the leakage of liquid
stools)
135.
Prevention
• Offer thechild high-fiber foods. such as fruits,
vegetables, beans, and whole-grain cereals
• Encourage the child to drink plenty of fluids.
Water is often the best.
• Promote physical activity. Regular physical
activity helps stimulate normal bowel function.
• Create a toilet routine. Regularly set aside time
after meals for the child to use the toilet
136.
continuation
• Remind thechild to heed nature's call. Some children get so
wrapped up in play that they ignore the urge to have a bowel
movement. If such delays occur often, they can contribute to
constipation.
• Be supportive. Reward the child's efforts, not results. Give
children small rewards for trying to move their bowels.
Possible rewards include stickers or a special book or game
that's only available after (or possibly during) toilet time. And
don't punish a child who has soiled his or her underwear.
• Review medications. If the child is taking a medication that
causes constipation, ask the doctor about other options.
Definition of distendedabdomen
• occurs when substances, such as air (gas) or
fluid, accumulate in the abdomen causing its
expansion.
• It is typically a symptom of an underlying
disease or dysfunction in the body, rather than
an illness in its own right.
140.
• Functional reasonsfor a distended abdomen
tend to involve digestive problems that cause
gas and/or digestive contents to accumulate.
Causes might include: Gas from functional
indigestion, food intolerances or irritable
bowel syndrome (IBS). Constipation causing a
build-up of feces and back-up of digestive
contents
141.
• Acute abdominaldistention in the pediatric
patient may be attributable to extraperitoneal
fluid, masses, organomegaly, air, an ileus, a
functional or mechanical bowel obstruction,
or injury and blood secondary to trauma