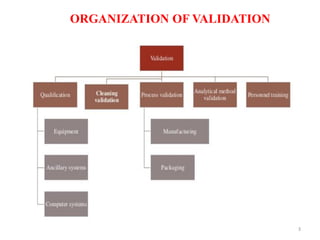
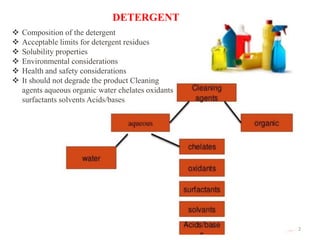
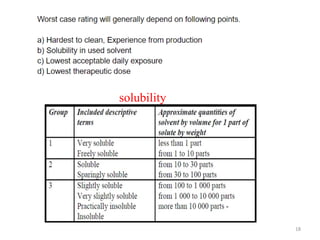
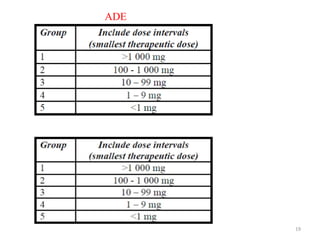
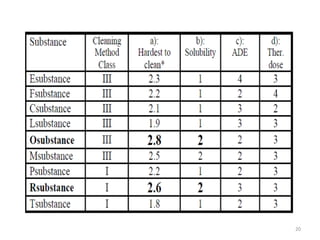
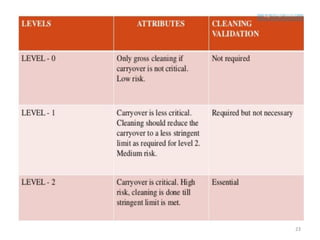
Cleaning validation is important to prove that equipment is consistently cleaned to acceptable levels to prevent contamination. There are three main goals of a cleaning validation program: 1) to prove equipment is consistently cleaned of product, detergent, and microbial residues; 2) to prevent possible contamination and cross-contamination; and 3) to demonstrate the cleaning process produces reproducible results. A cleaning validation program establishes standard cleaning procedures, validates the cleaning process, and provides documented evidence that the approved cleaning procedure is suitable for processing medicinal products.