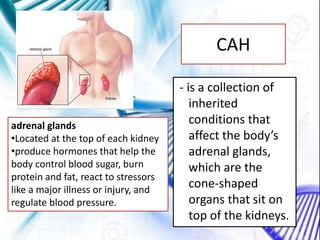
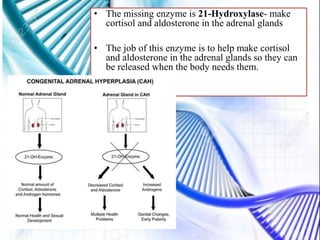
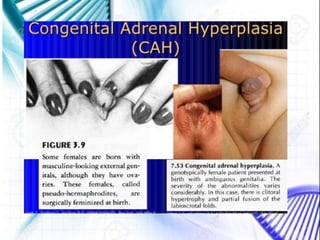
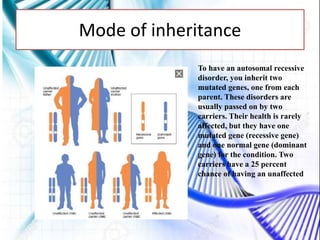
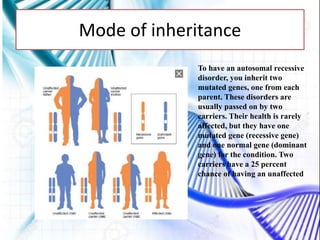
CAH and galactosemia are genetic disorders. CAH is caused by a deficiency in the enzyme 21-hydroxylase, resulting in enlarged adrenal glands and excess production of male sex hormones. It can cause genital anomalies in females and salt-wasting issues. Galactosemia is caused by a deficiency in the GALT enzyme, preventing the breakdown of galactose which can lead to issues like cataracts if untreated. Both require dietary management to avoid complications.