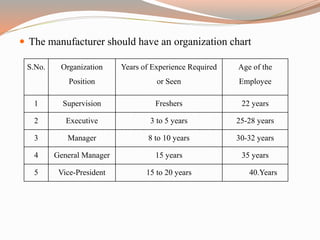
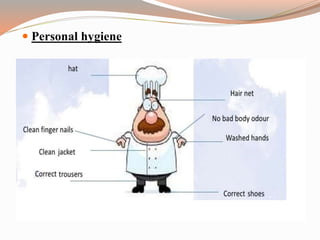
The document provides a comprehensive literature review on current Good Manufacturing Practices (cGMP) in the pharmaceutical industry, explaining its significance in ensuring product quality and compliance with legal standards. It outlines the roles of various regulatory authorities and details elements such as production controls, personnel organization, and equipment requirements to maintain quality assurance. cGMP is defined as a system that guarantees products are consistently produced and controlled to meet quality standards necessary for their intended use.