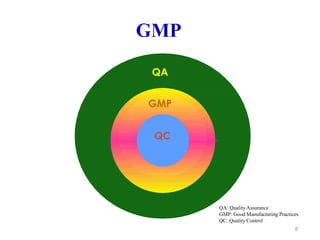
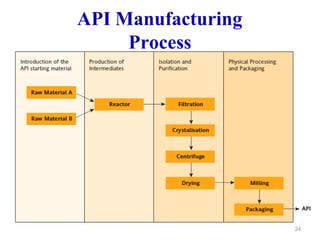
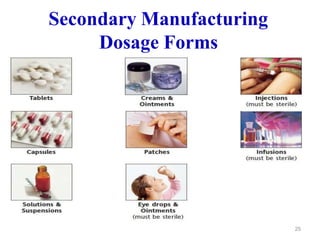
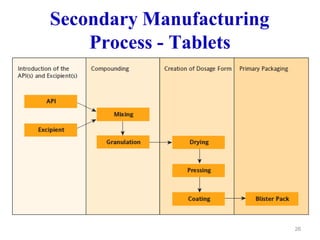
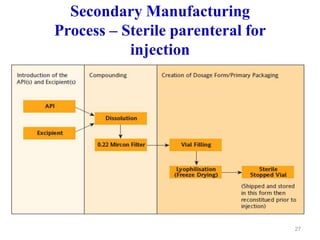
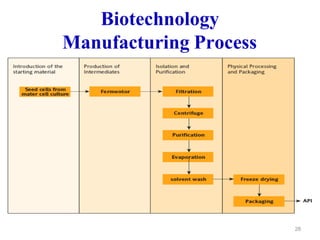
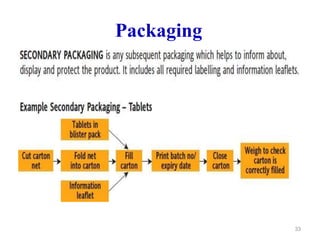
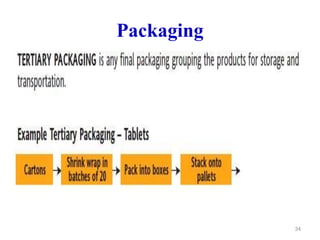
The document discusses Good Manufacturing Practices (GMP) which are a system for ensuring that products are consistently produced and controlled according to quality standards. It covers GMP guidelines from various organizations, key aspects of GMP like packaging and facilities, and concepts like quality assurance, quality control, documentation practices. GMP is important for minimizing risks in pharmaceutical production and ensuring medicine quality and safety. Adherence to GMP regulations is necessary for pharmaceutical manufacturers and exporters.