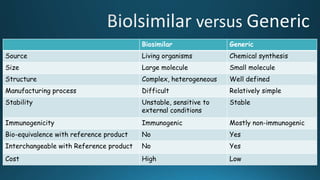
The document discusses the development of biosimilars as a cost-effective alternative to biologics, which are sophisticated and expensive drugs derived from living organisms. It defines biosimilars as similar biological products that demonstrate quality, safety, and efficacy comparable to reference biologics but are not chemically identical. The regulatory framework for their approval includes stringent guidelines and requires extensive studies to ensure safety and effectiveness, particularly in terms of immunogenicity and pharmacokinetics.