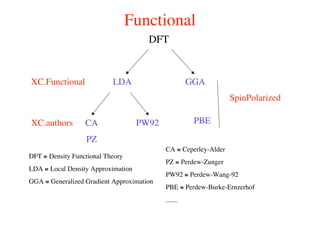
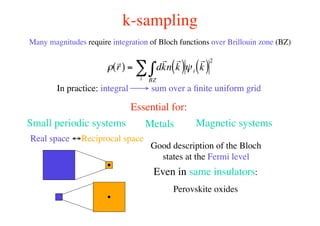
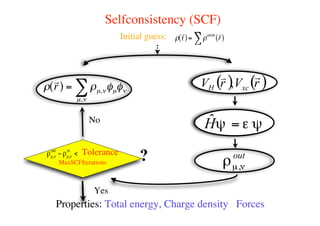
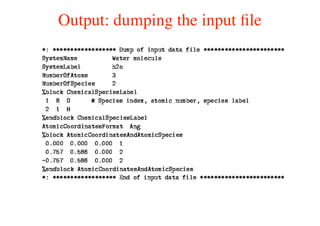
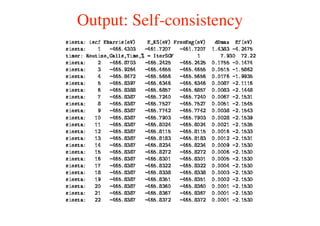
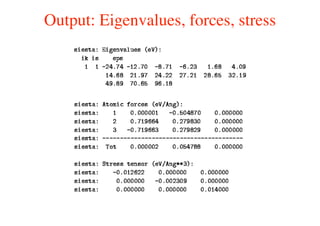
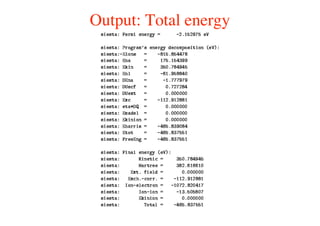
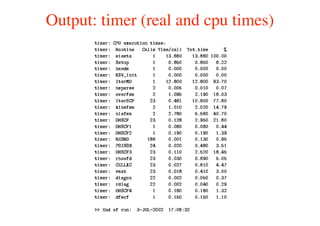
The document provides an introduction to running the Siesta software package for performing density functional theory (DFT) calculations. It describes the basic input variables needed, including system descriptors, structural parameters, functional and basis set specifications. It also outlines how to run Siesta from the command line and analyze outputs such as electronic band structures, densities of states, and charge densities. Post-processing tools are also summarized.