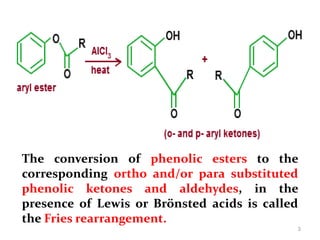
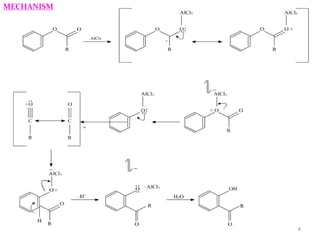
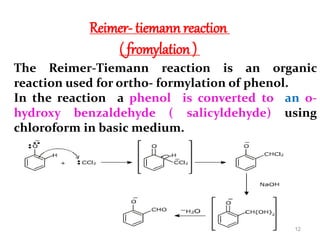
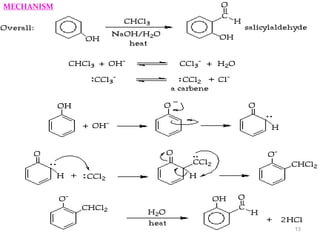
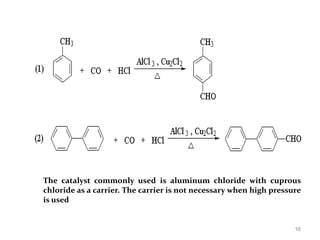
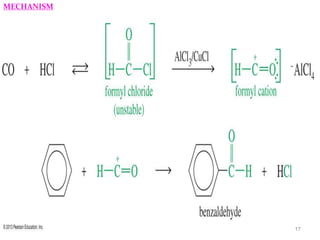
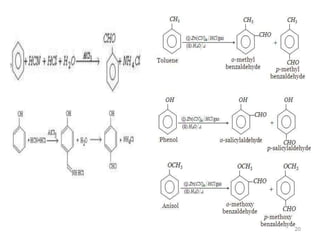
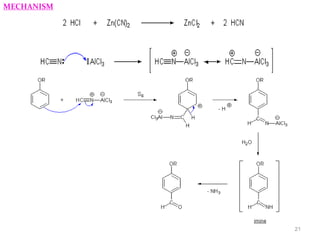
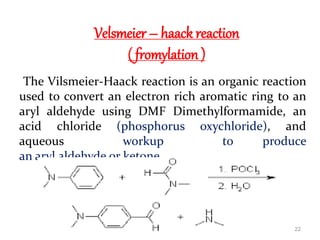
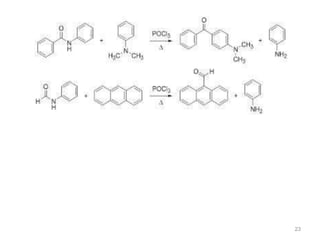
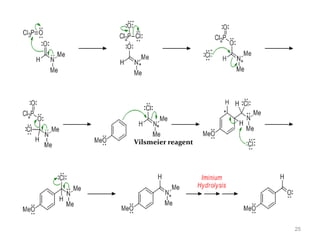
The document discusses several aromatic rearrangement reactions including the Fries rearrangement, photo-Fries rearrangement, Reimer-Tiemann reaction, Gattermann-Koch reaction, Gattermann aldehyde synthesis, and Vilsmeier-Haack reaction. The Fries rearrangement involves the rearrangement of phenolic esters to ortho- and para-substituted phenolic ketones/aldehydes using Lewis or Brønsted acids. The photo-Fries rearrangement is similar but uses light instead of acids. The Reimer-Tiemann, Gattermann-Koch, and Vilsmeier-Haack reactions all involve the formylation of aromatic compounds to produce





![There are two main variants of the Fries
rearrangement:
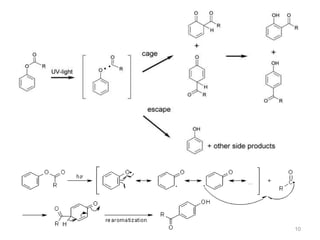
A. Upon irradiation with light phenolic esters
undergo the same transformation, which is
known as the photo-Fries rearrangement.
B. An anionic ortho-Fries rearrangement takes
place when ortho-lithiated O-aryl carbamates
undergo a facile intramolecular [1,3]-acyl
migration to give substituted salicylamides at
room temperature.
7](https://image.slidesharecdn.com/aromaticrearrangements-200420080501/85/Aromatic-rearrangements-7-320.jpg)






























![Cycloaddition reactions [2+2]](https://cdn.slidesharecdn.com/ss_thumbnails/cycloadditionreactions22-200514145354-thumbnail.jpg?width=640&height=640&fit=bounds)




















































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)










