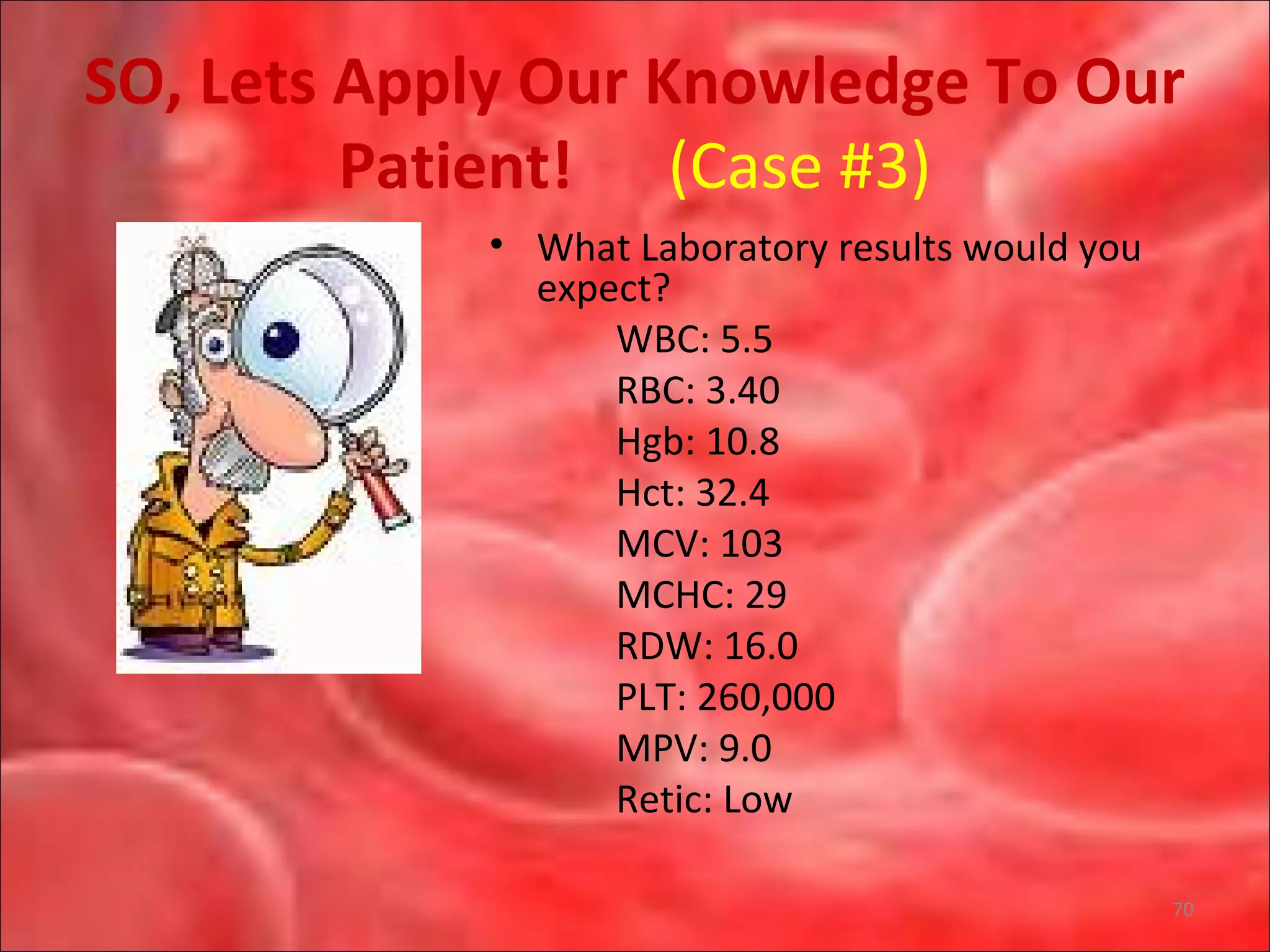
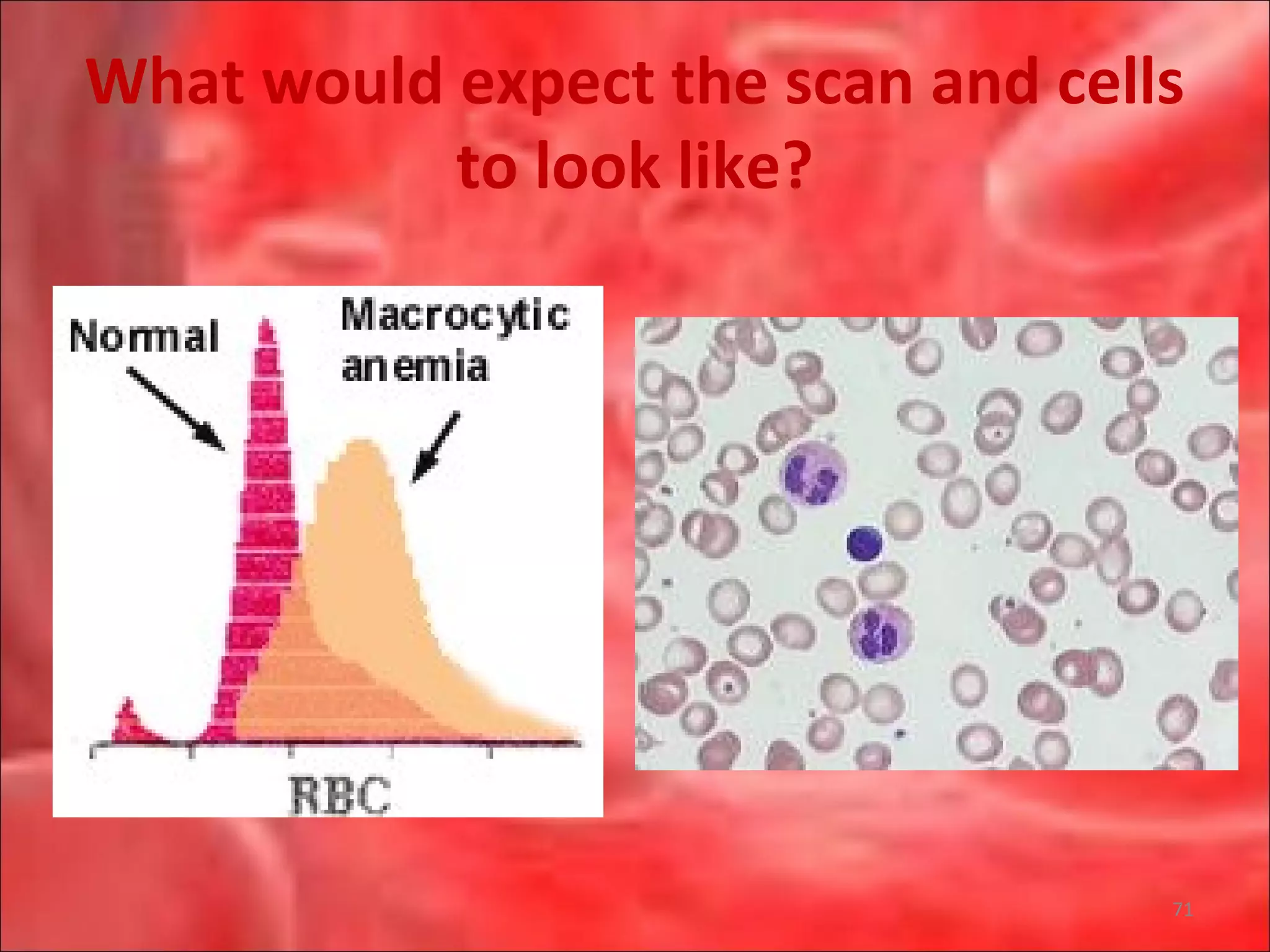
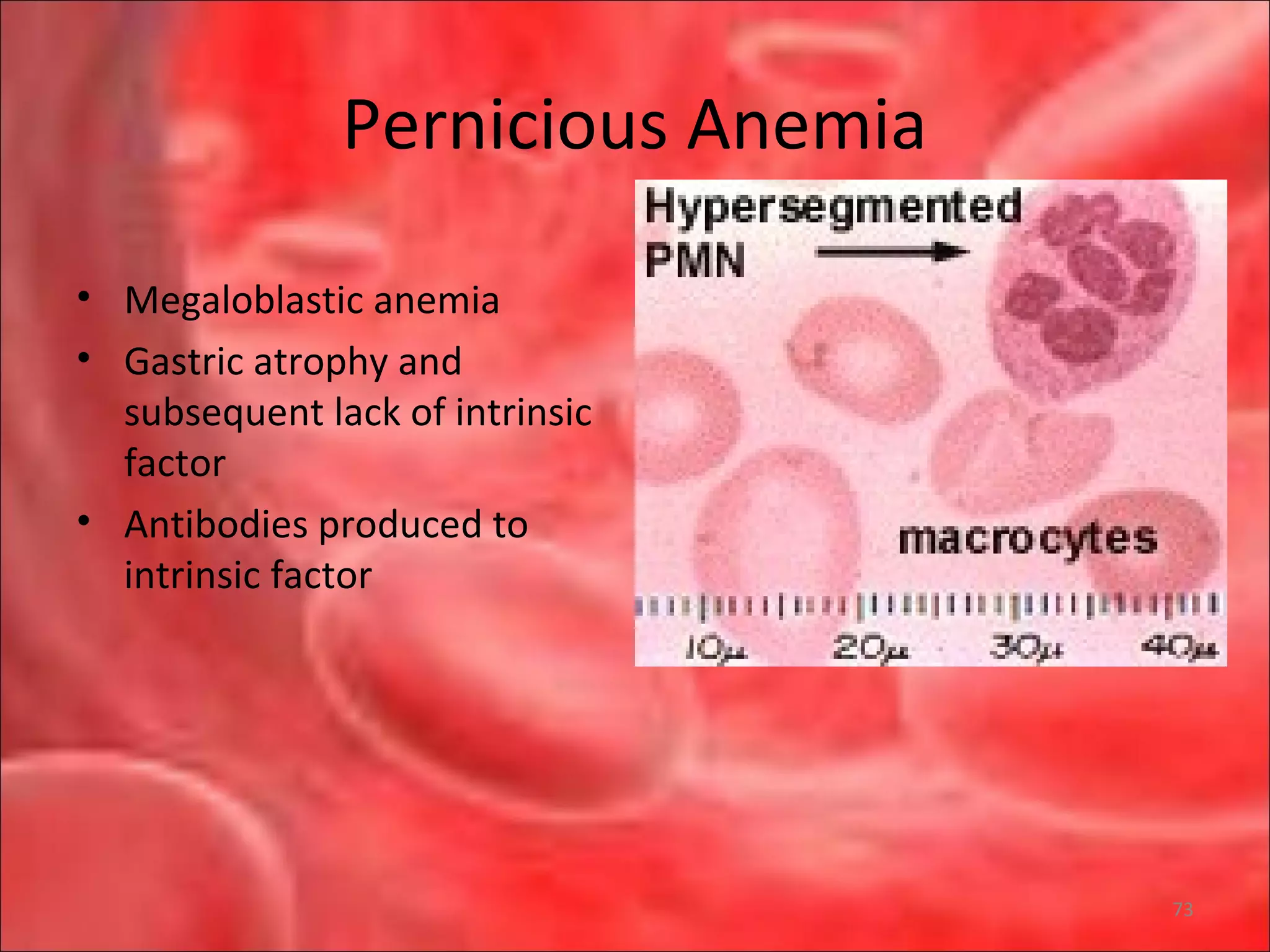
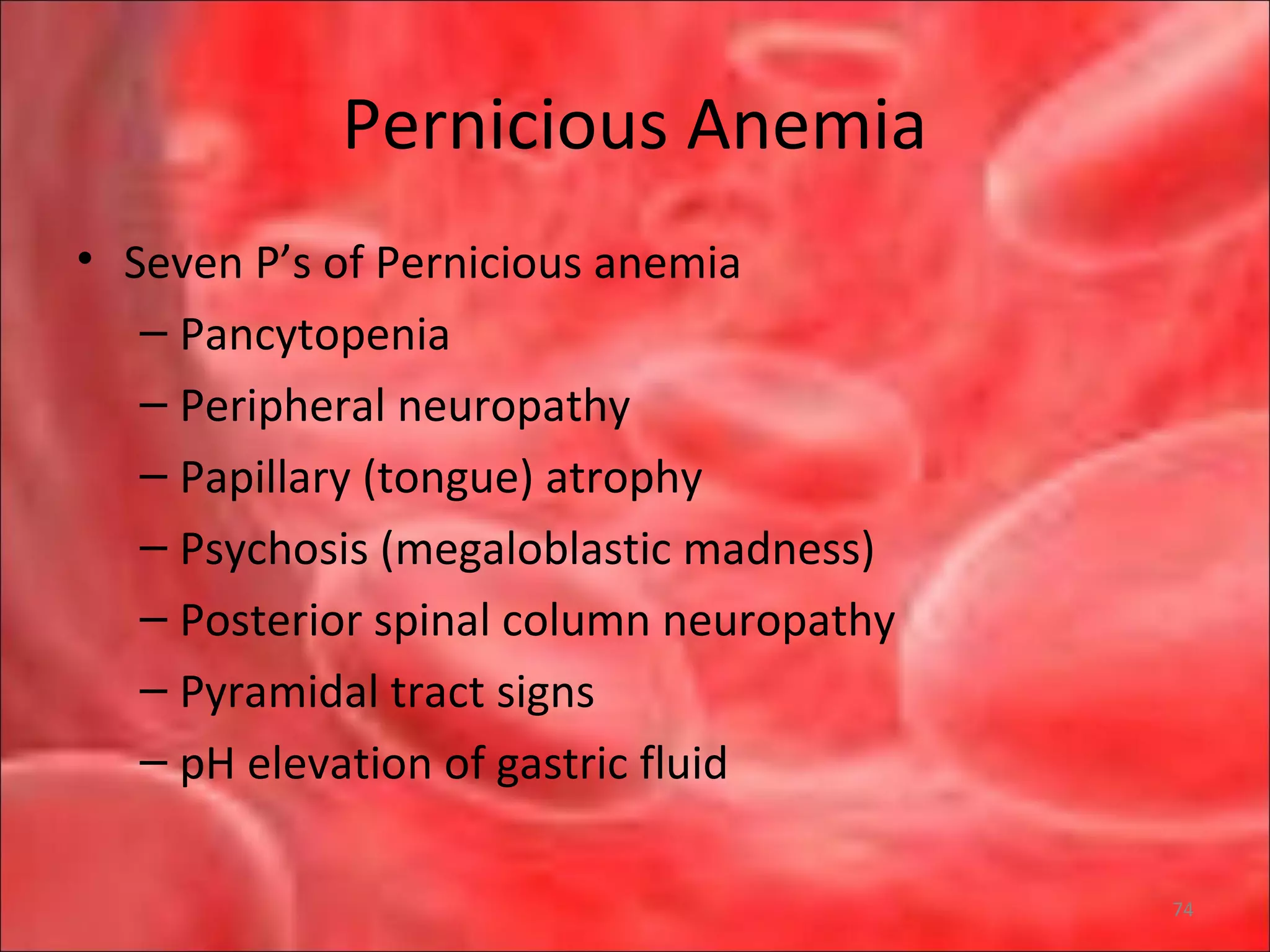
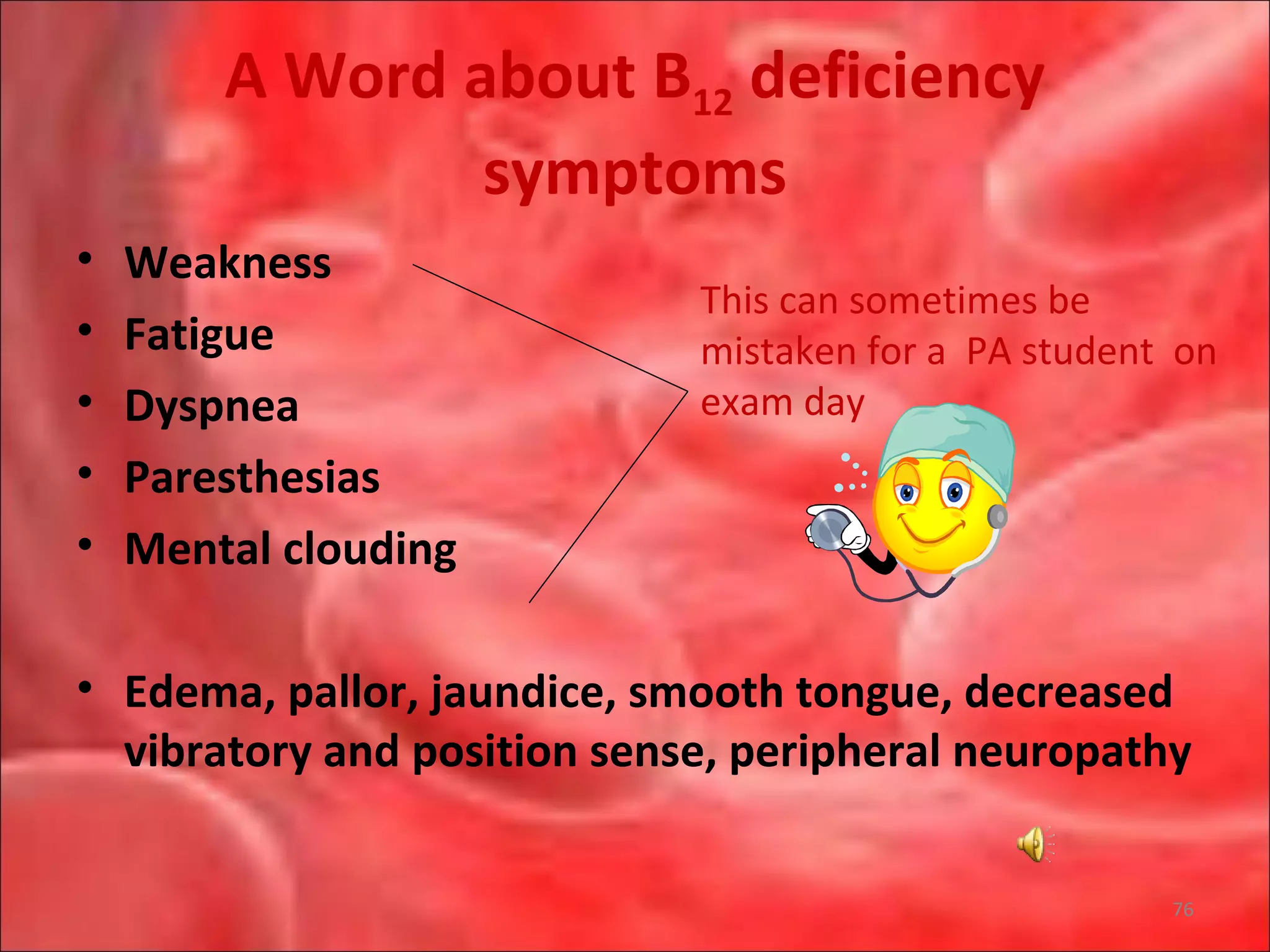
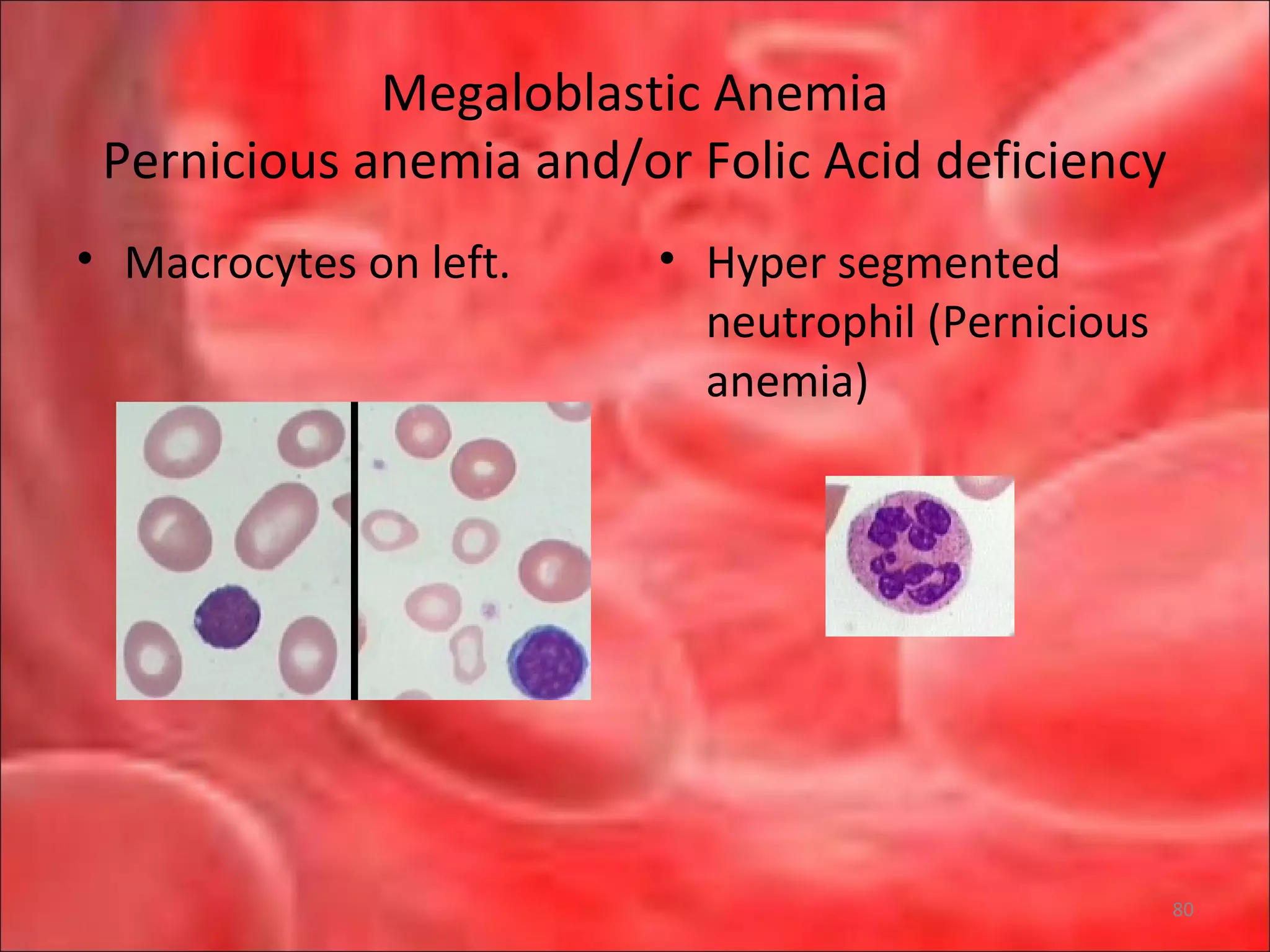
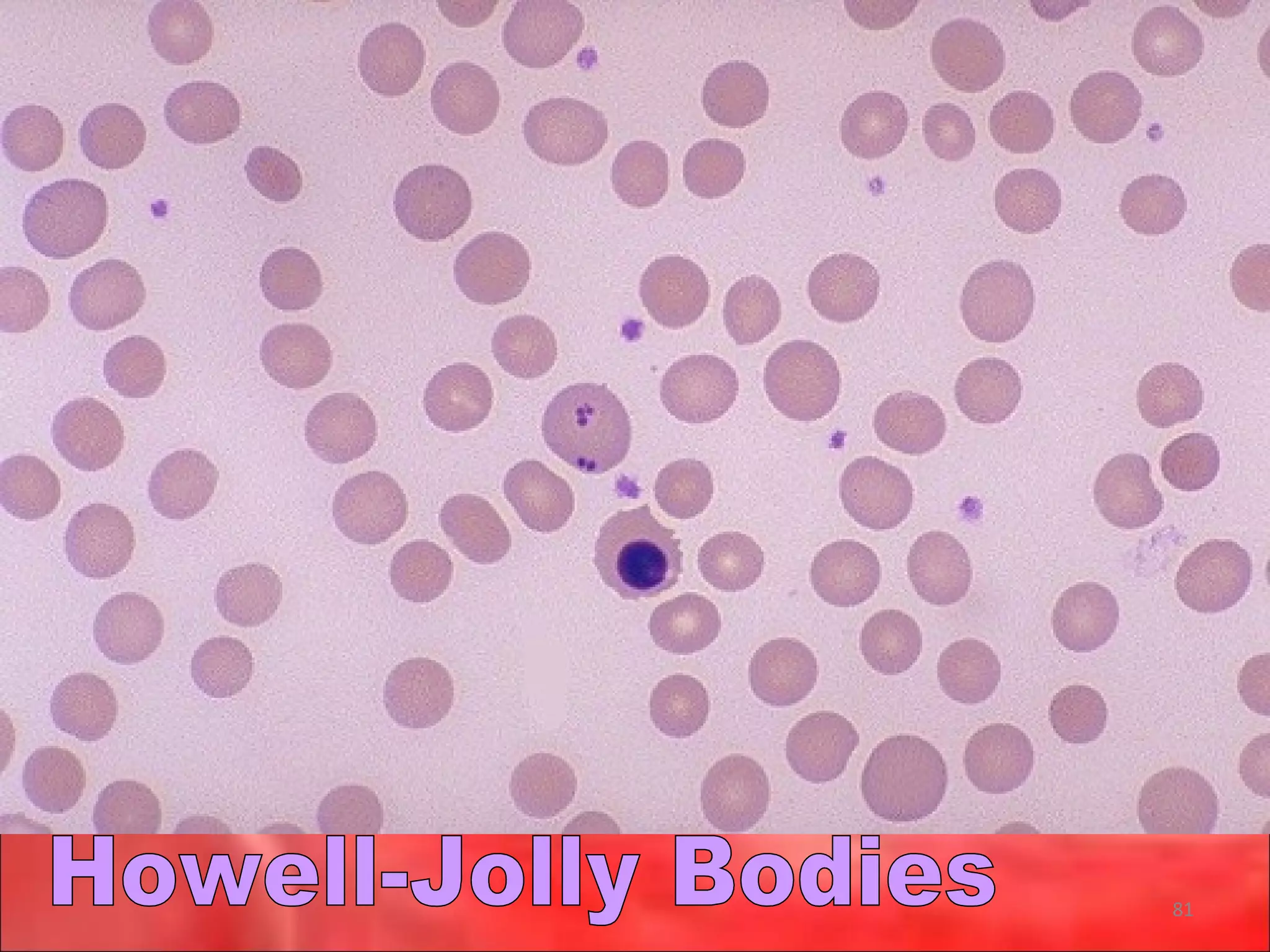
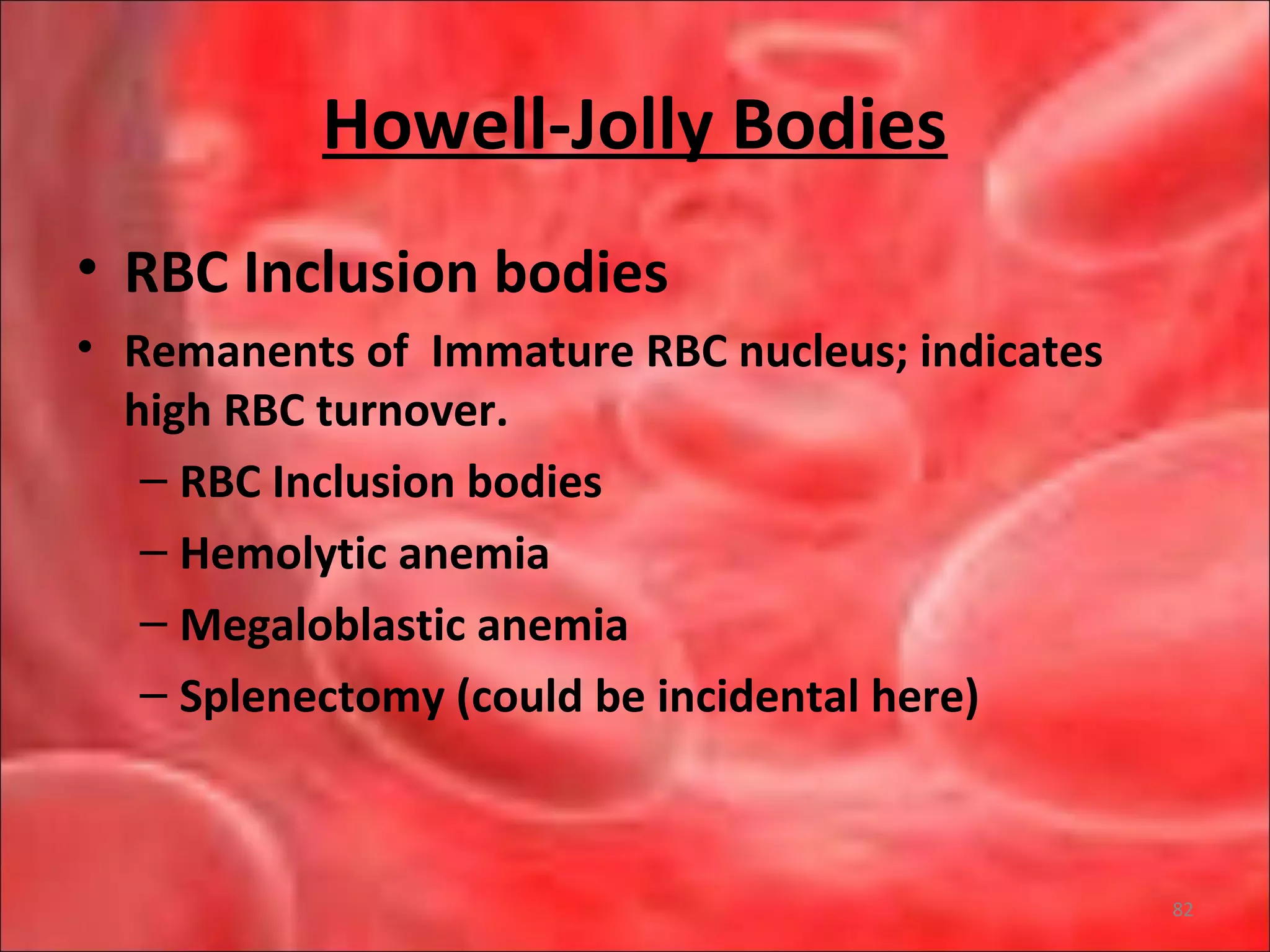
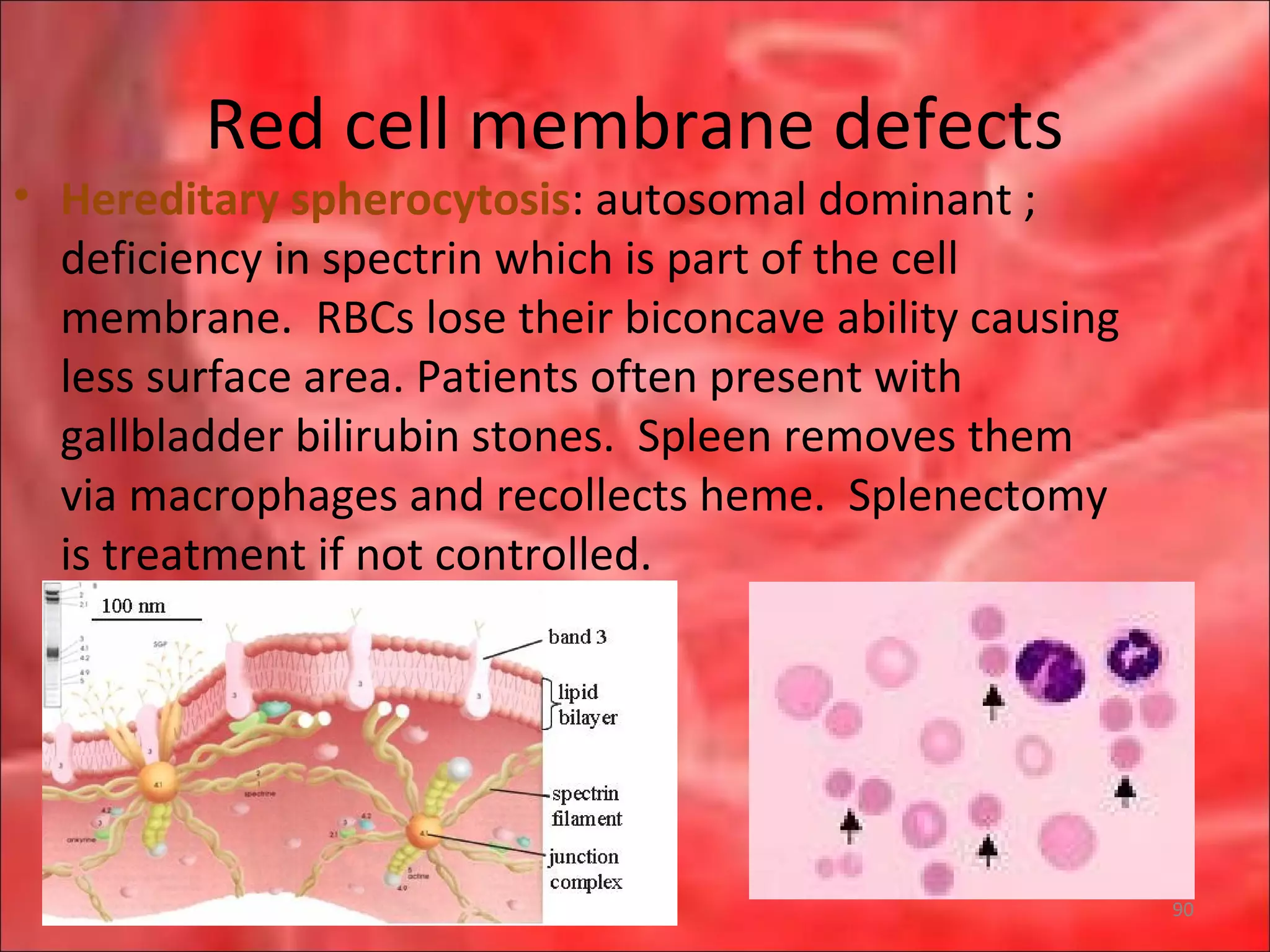
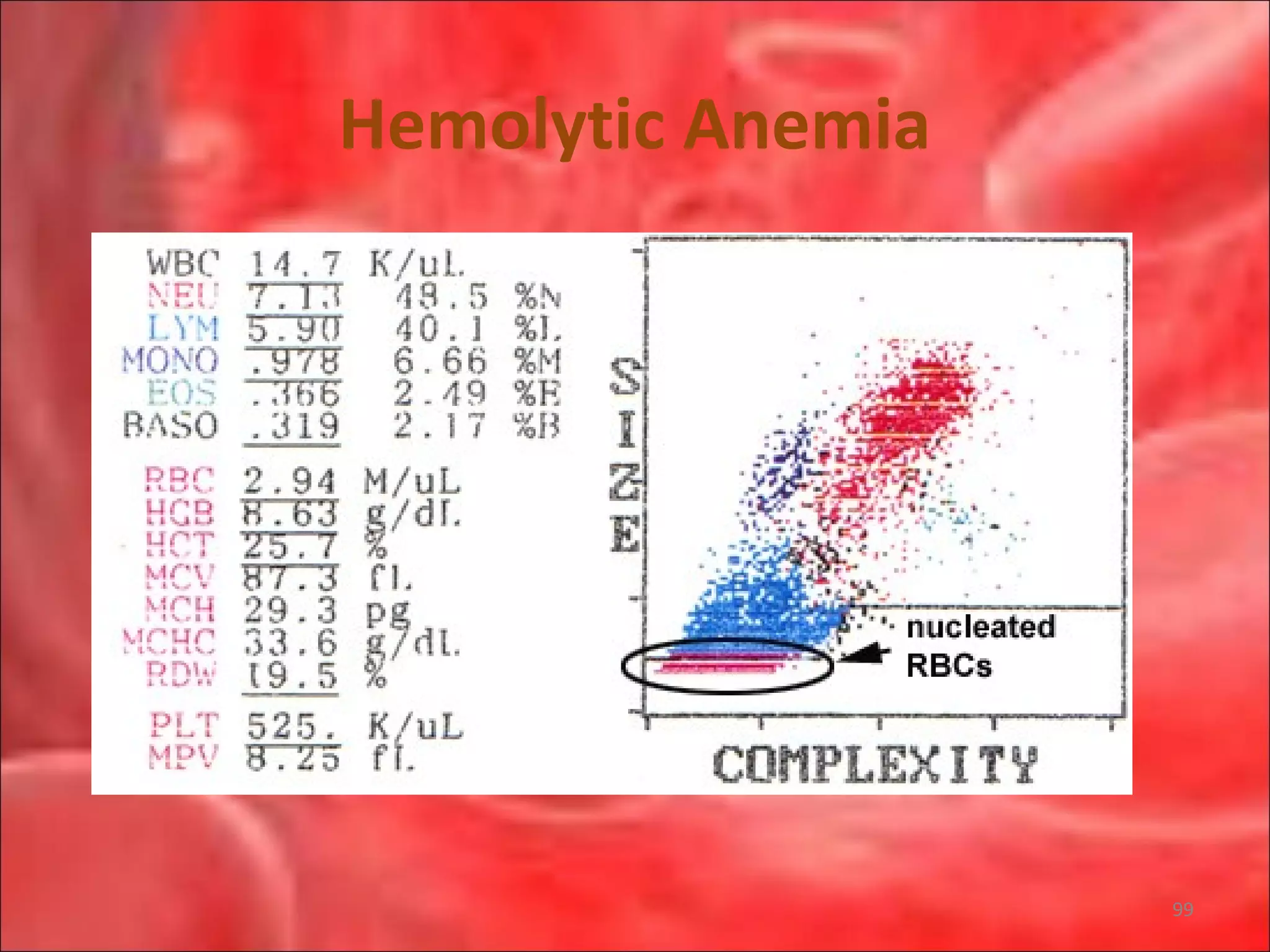
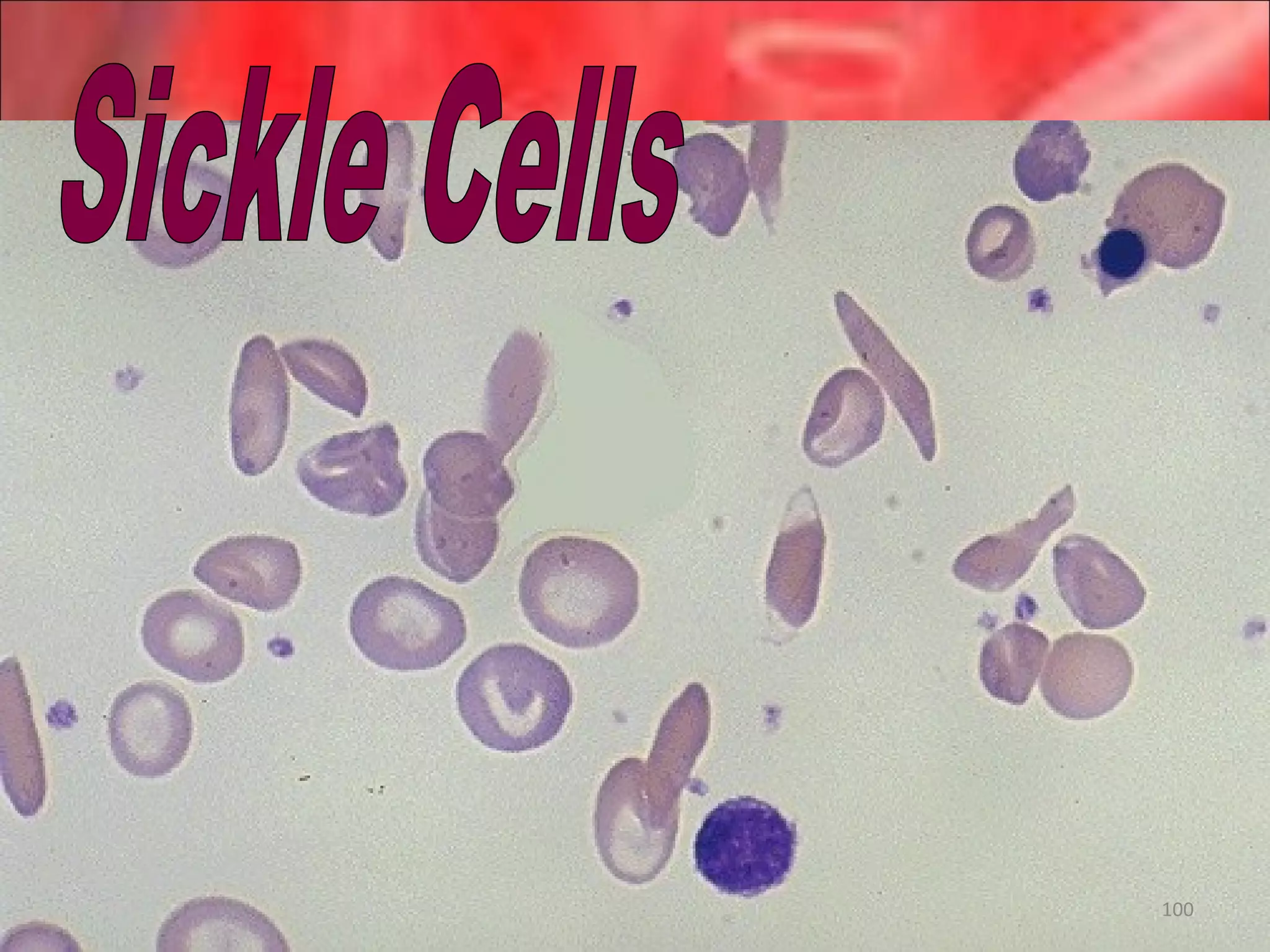
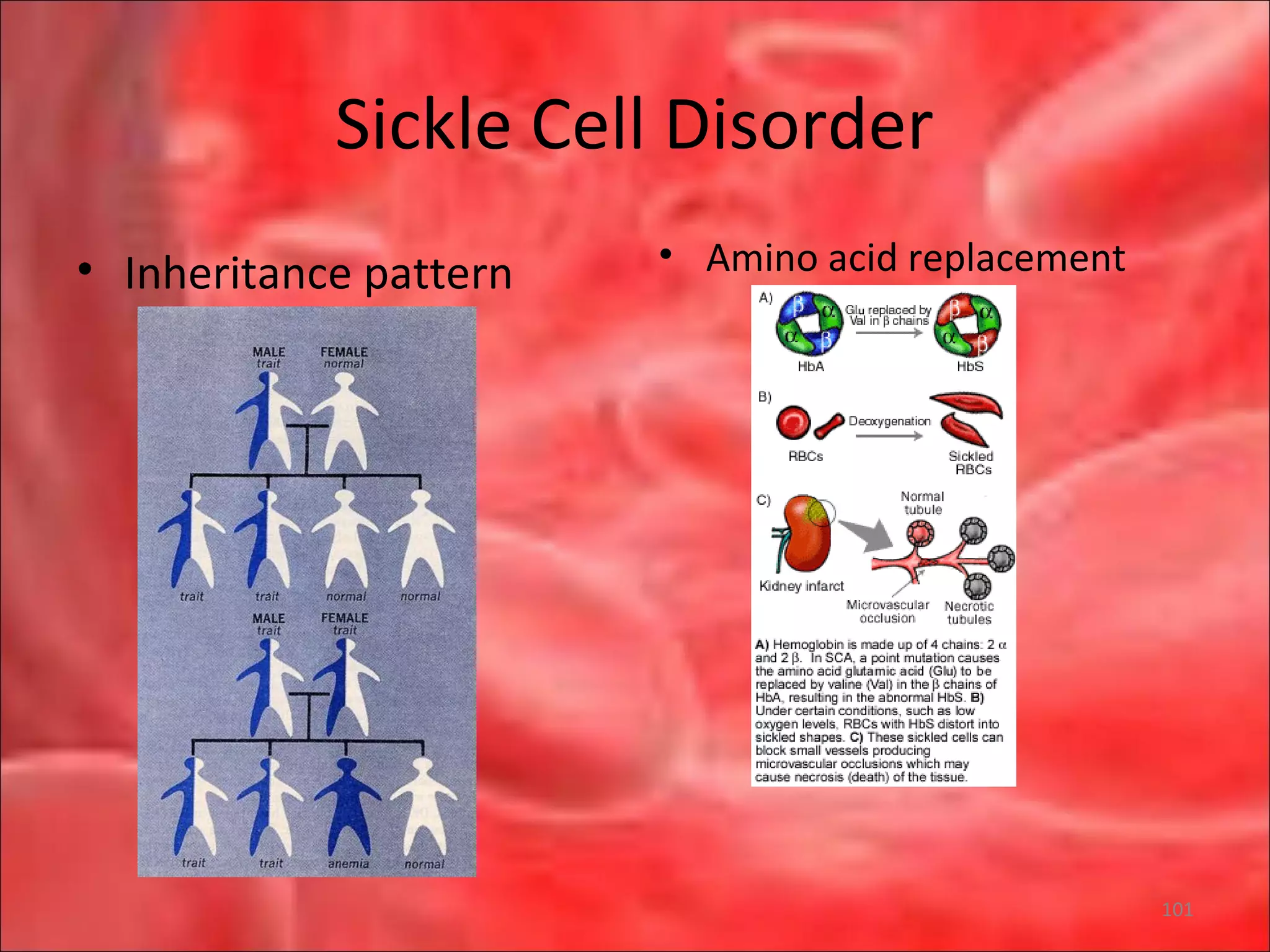
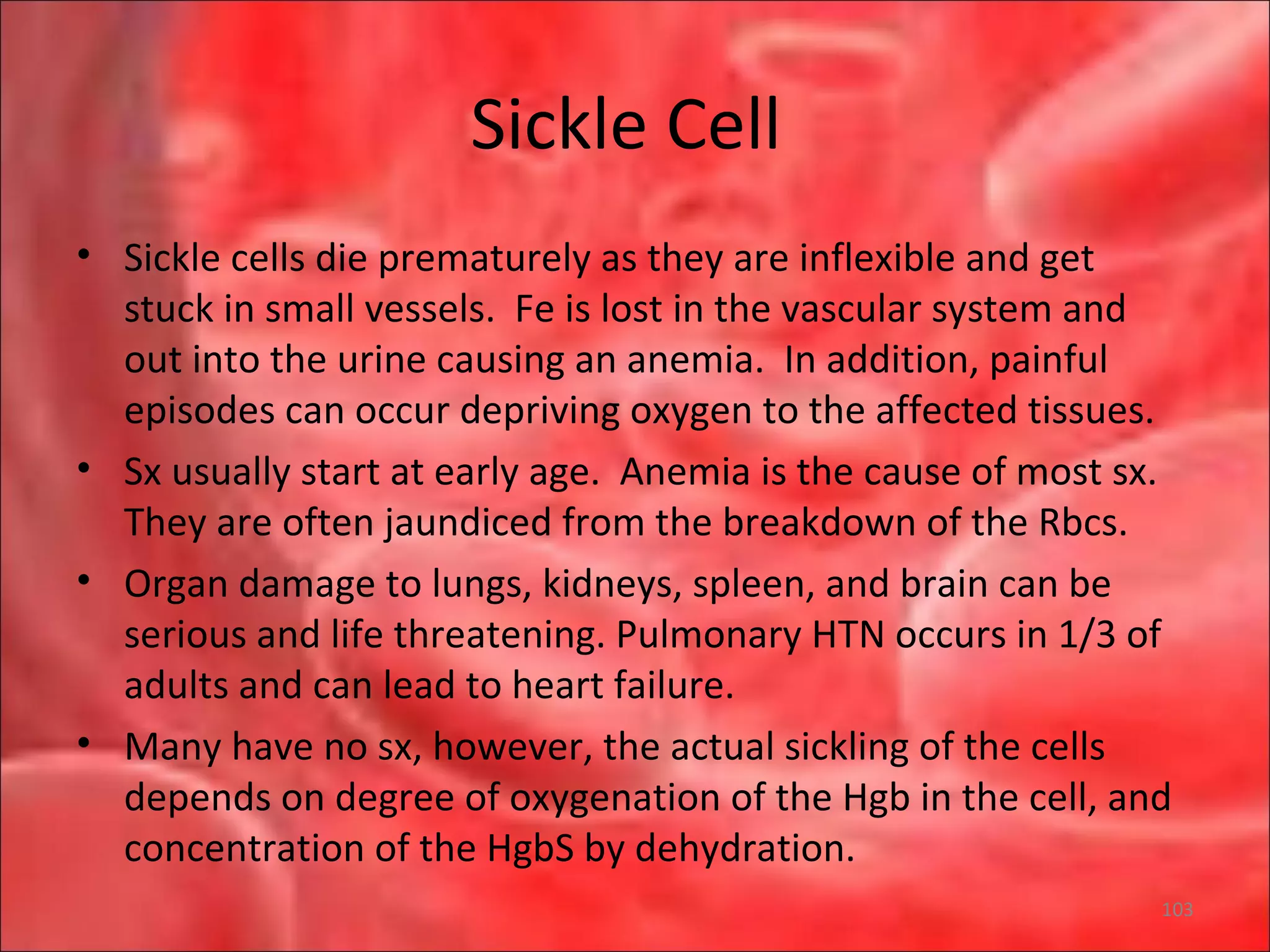
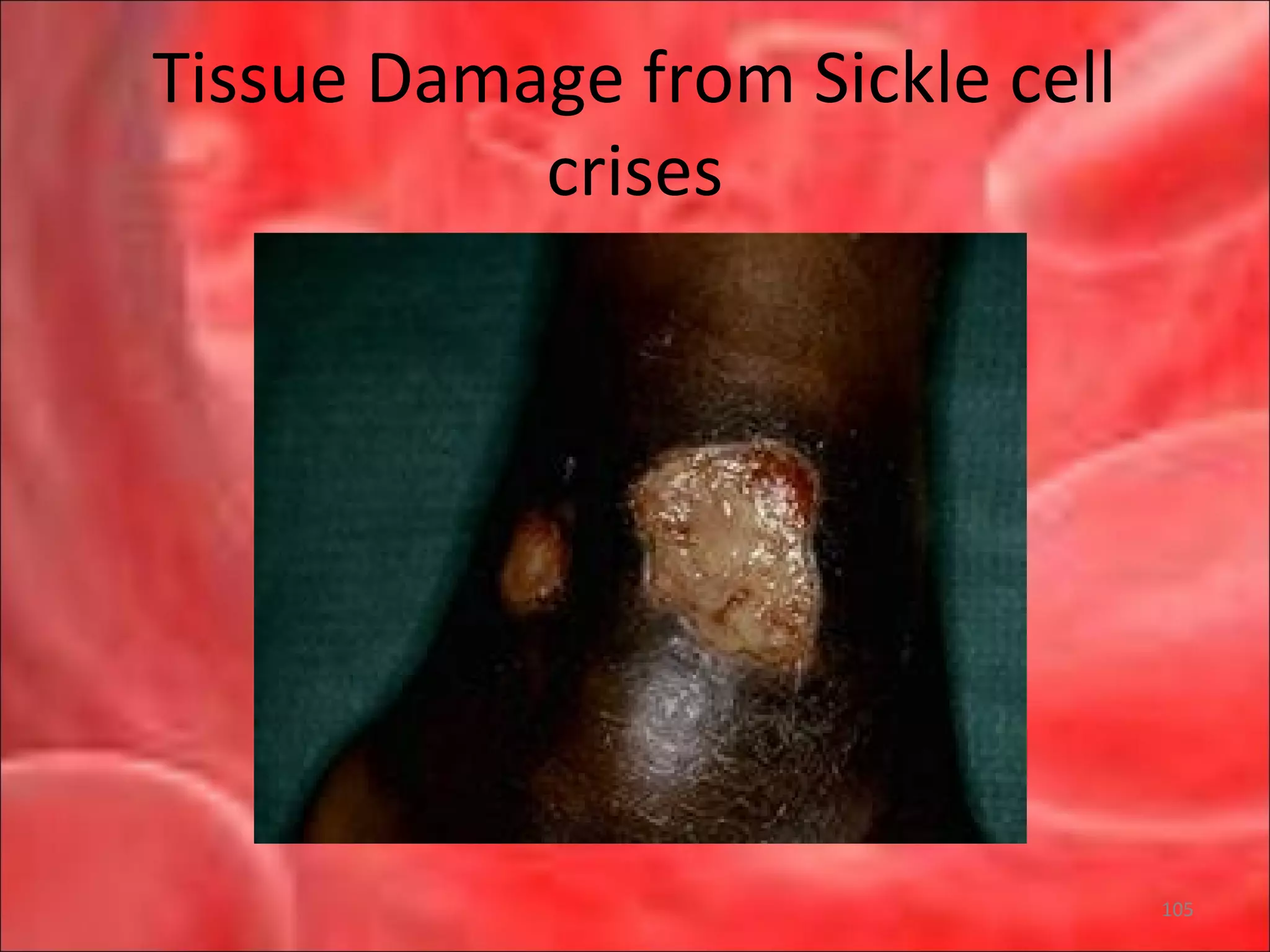
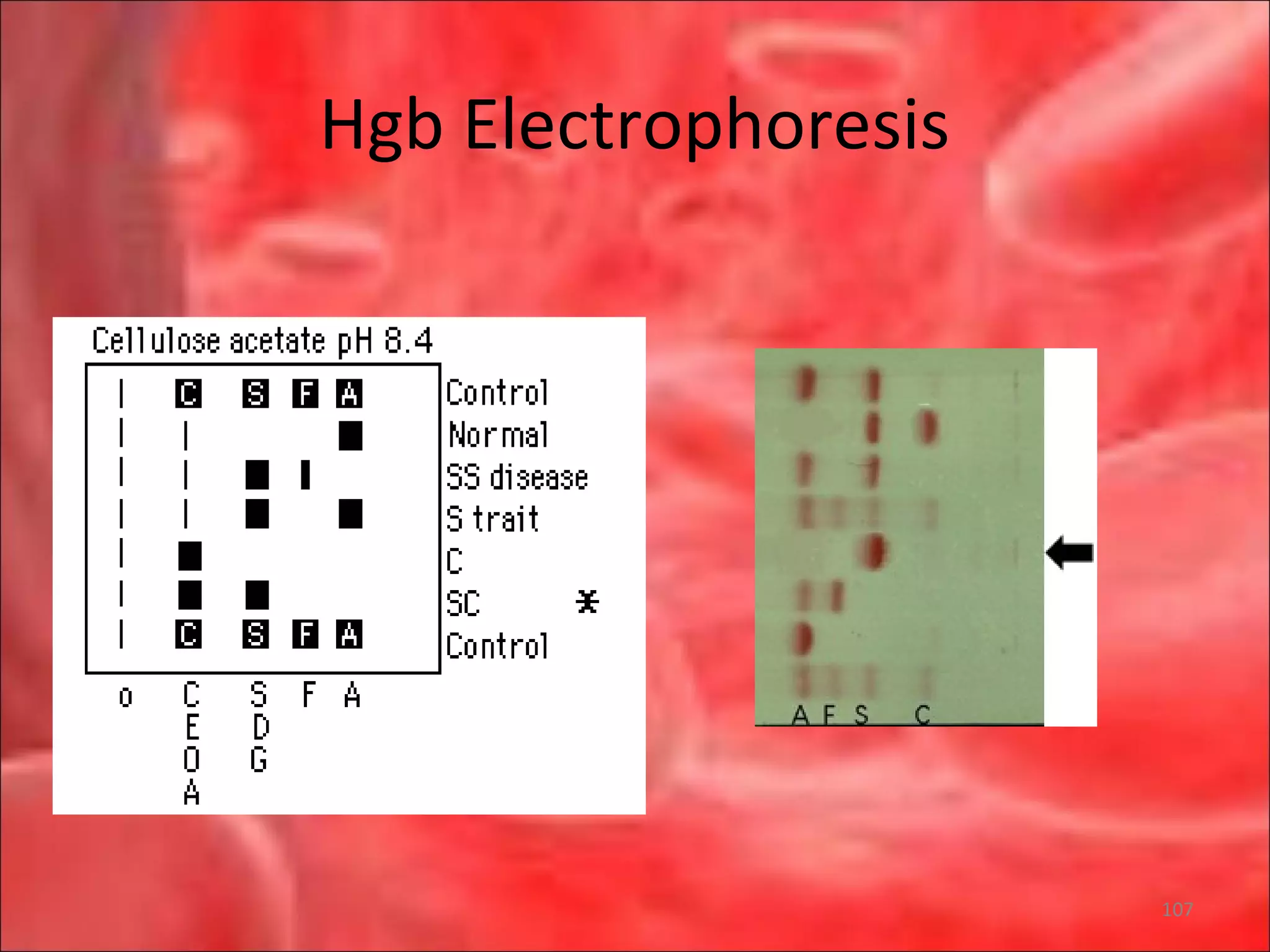
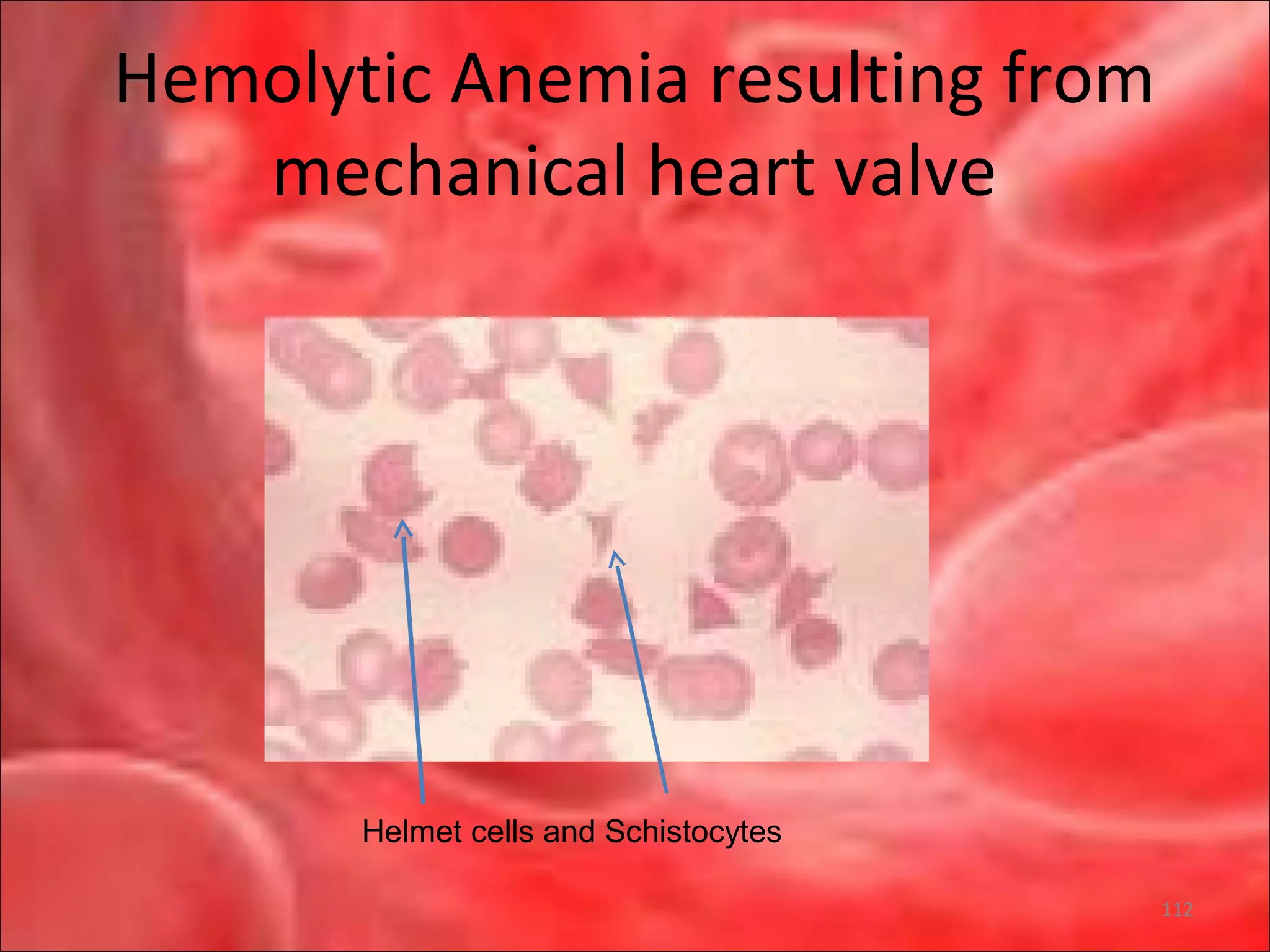
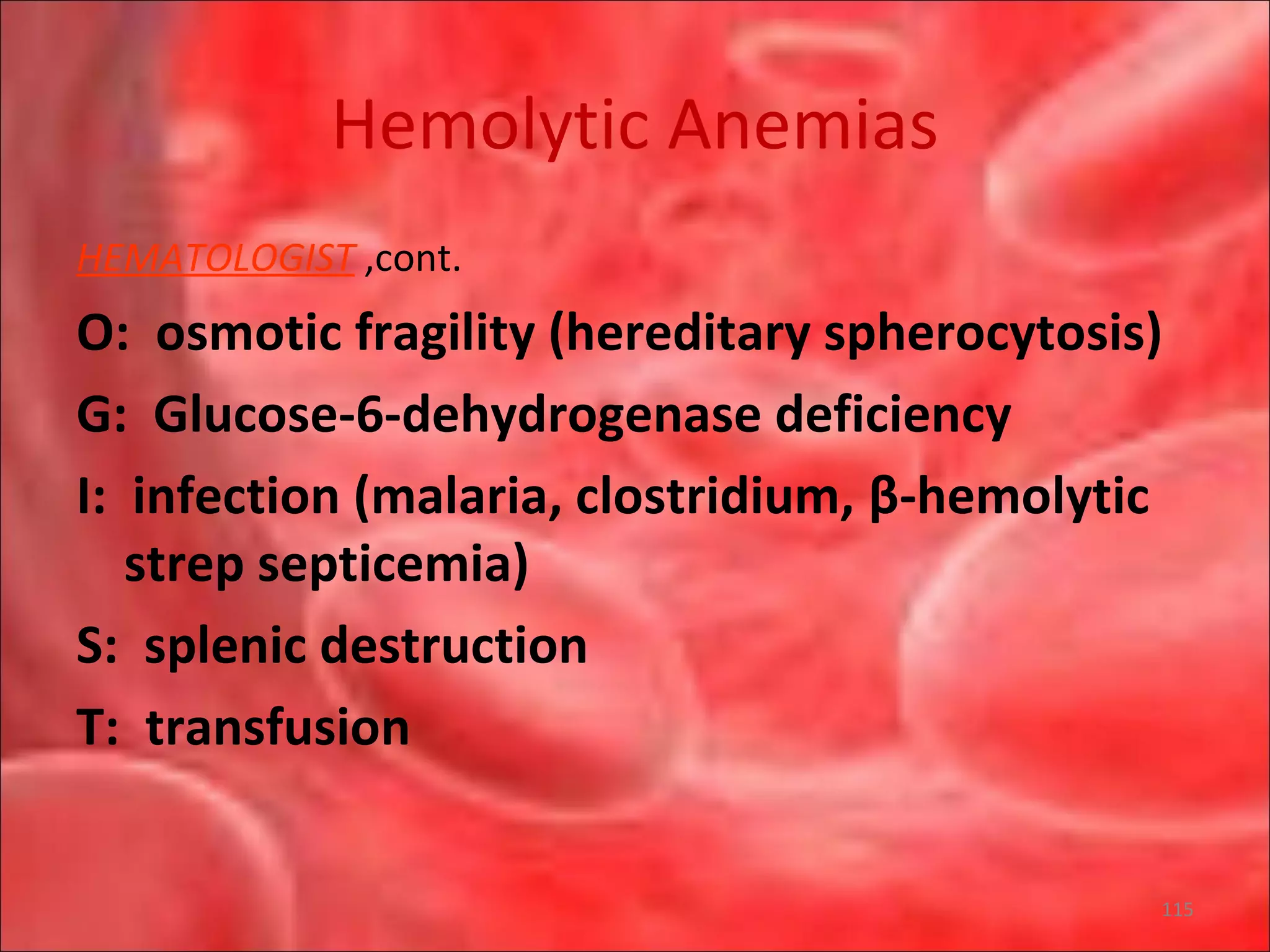
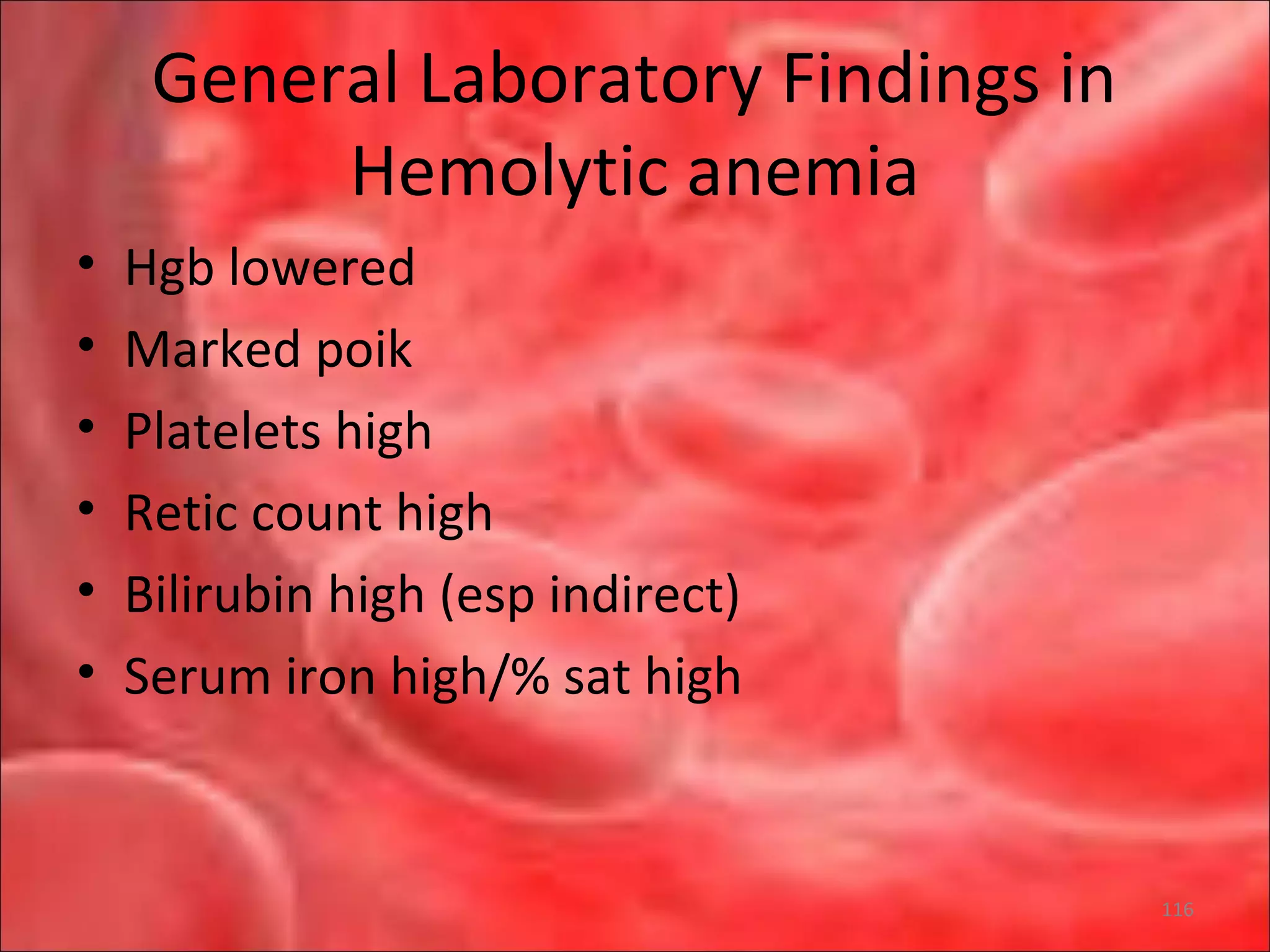
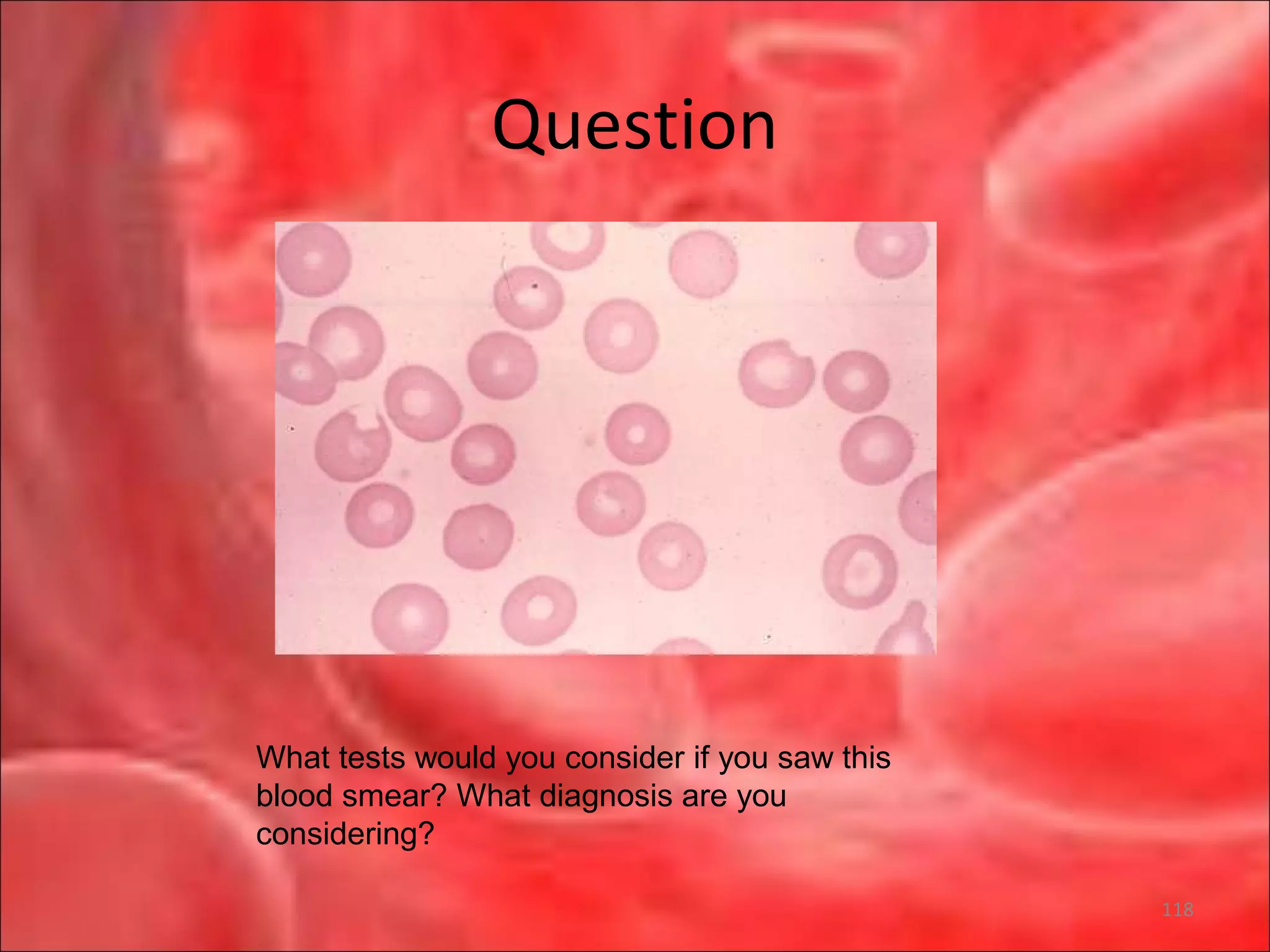
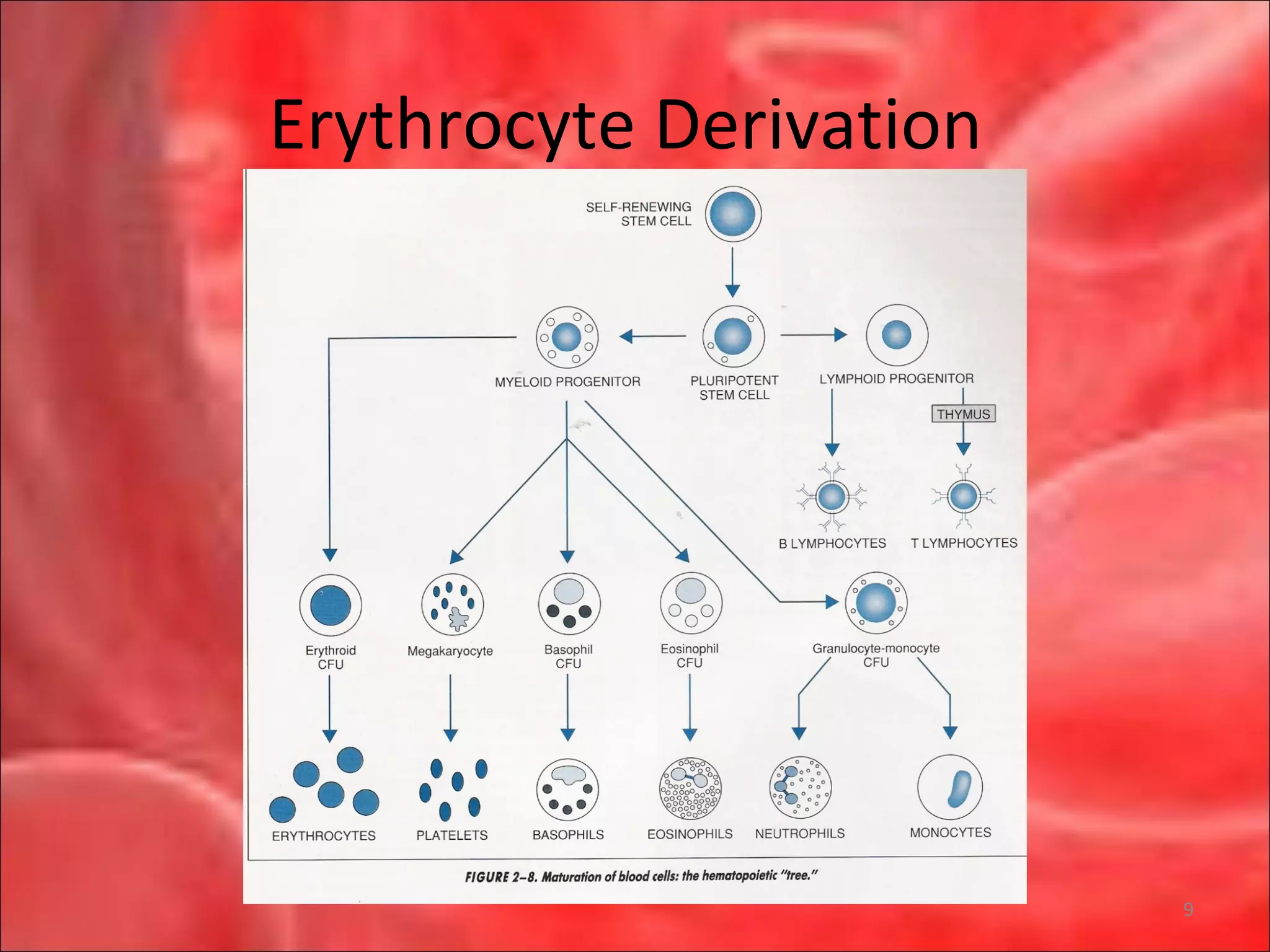
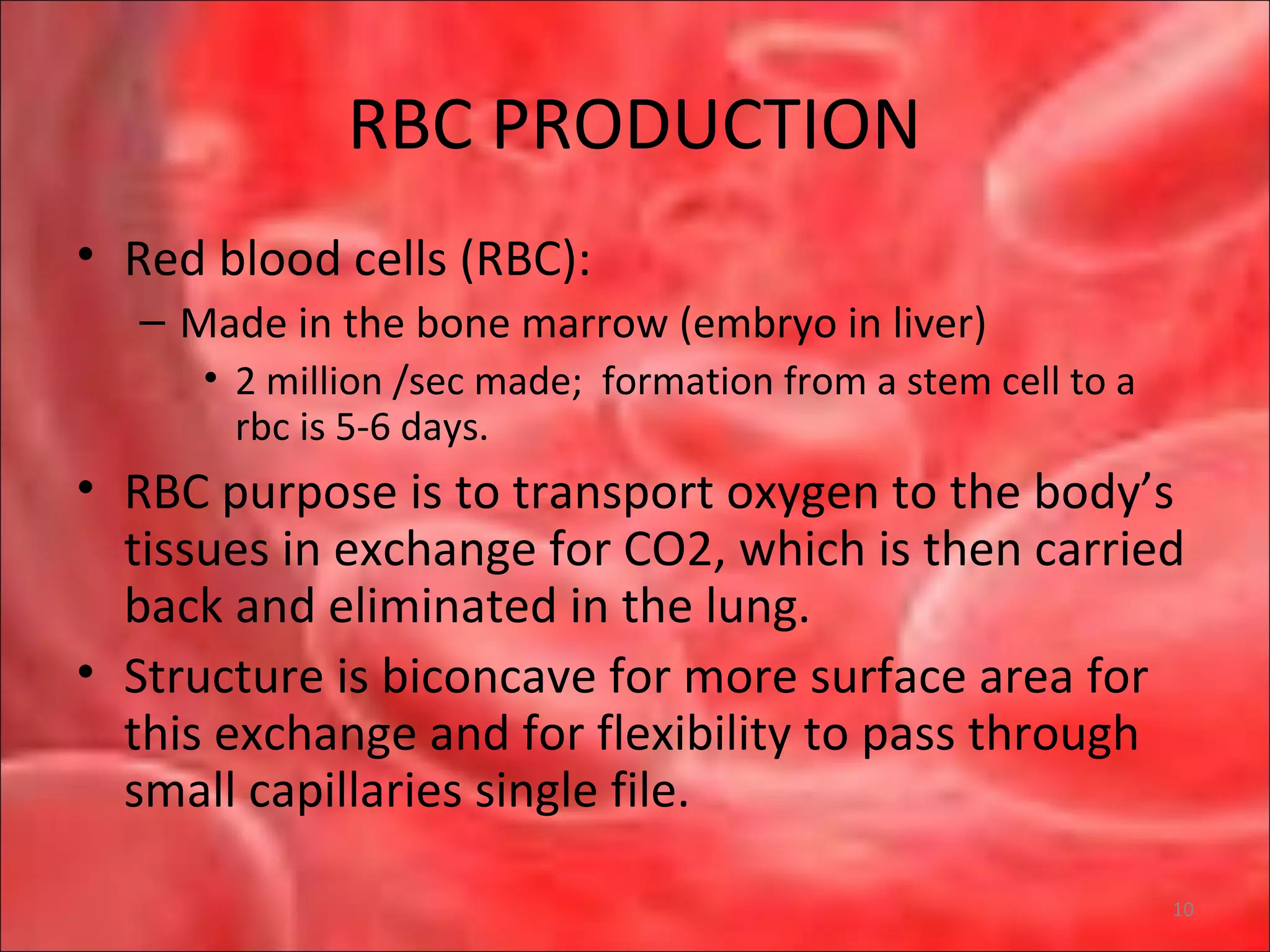
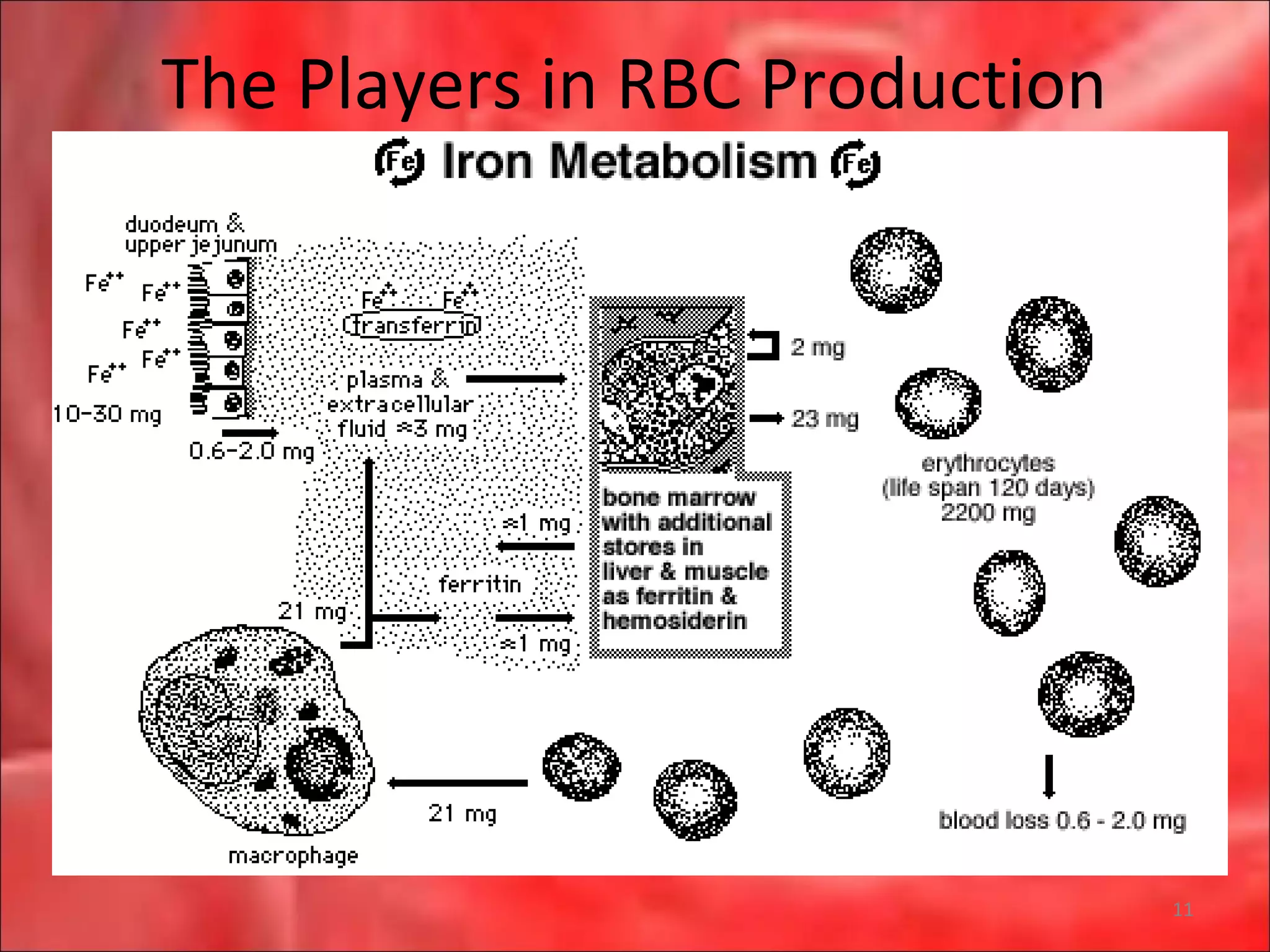
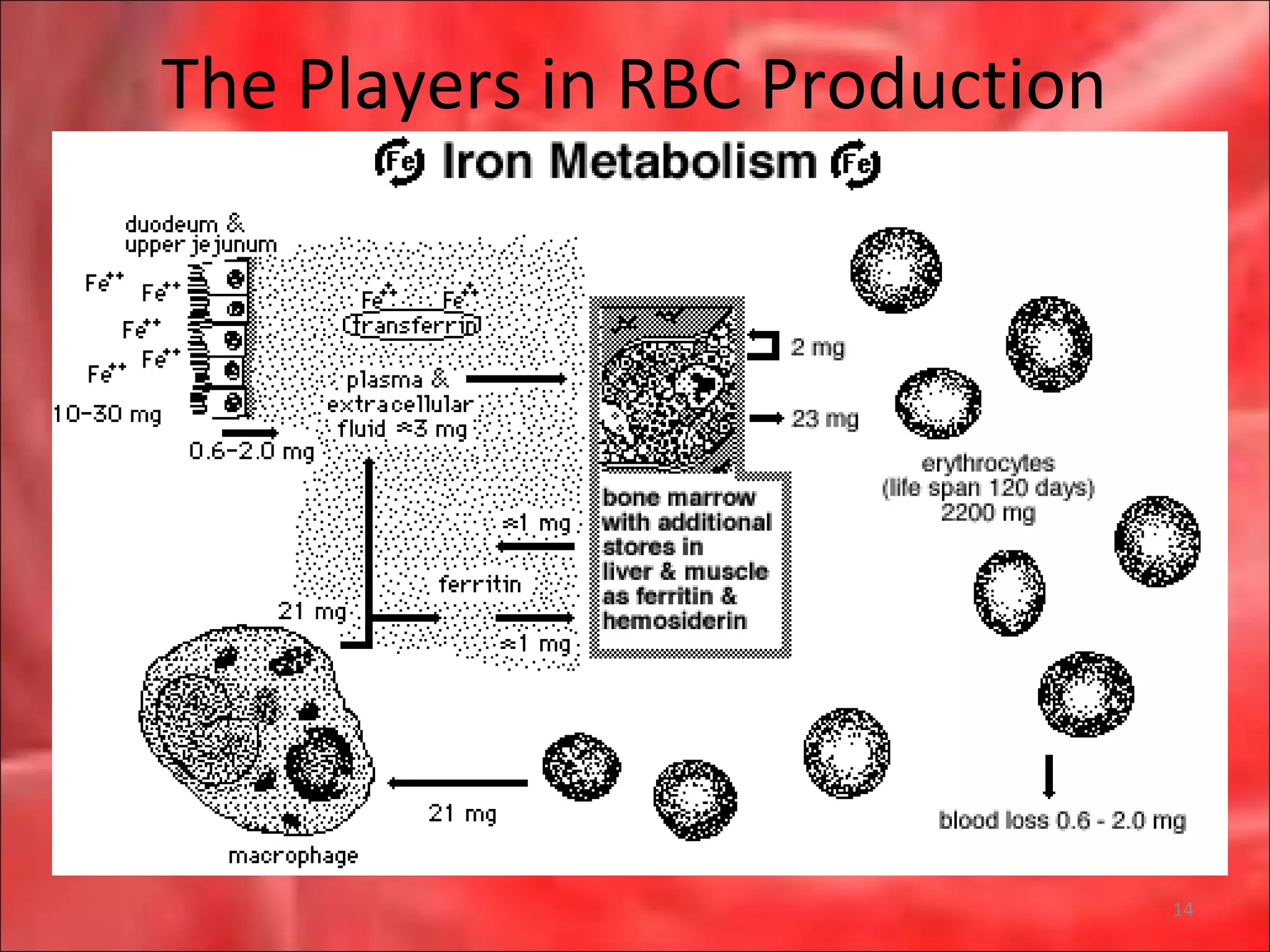
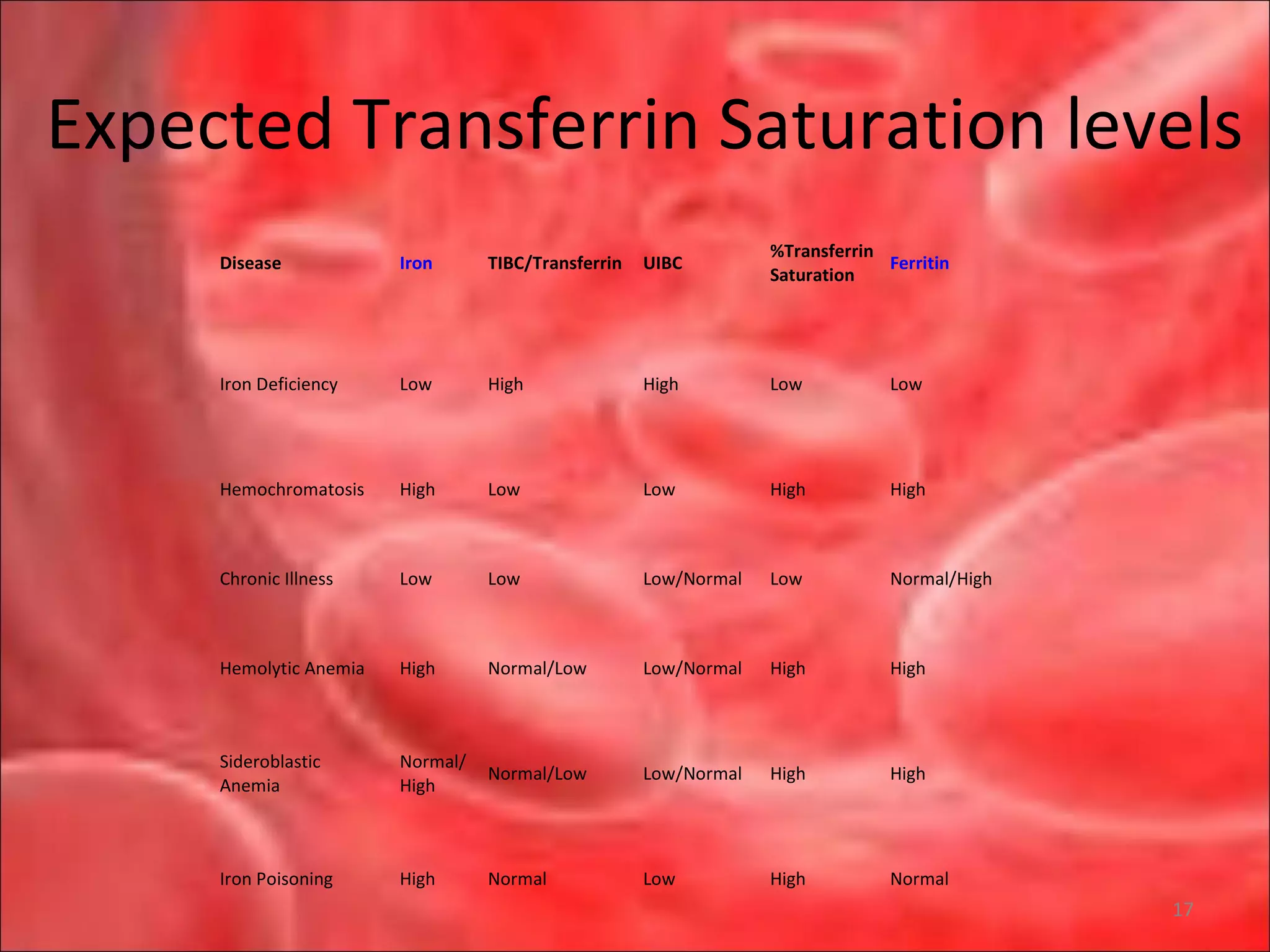
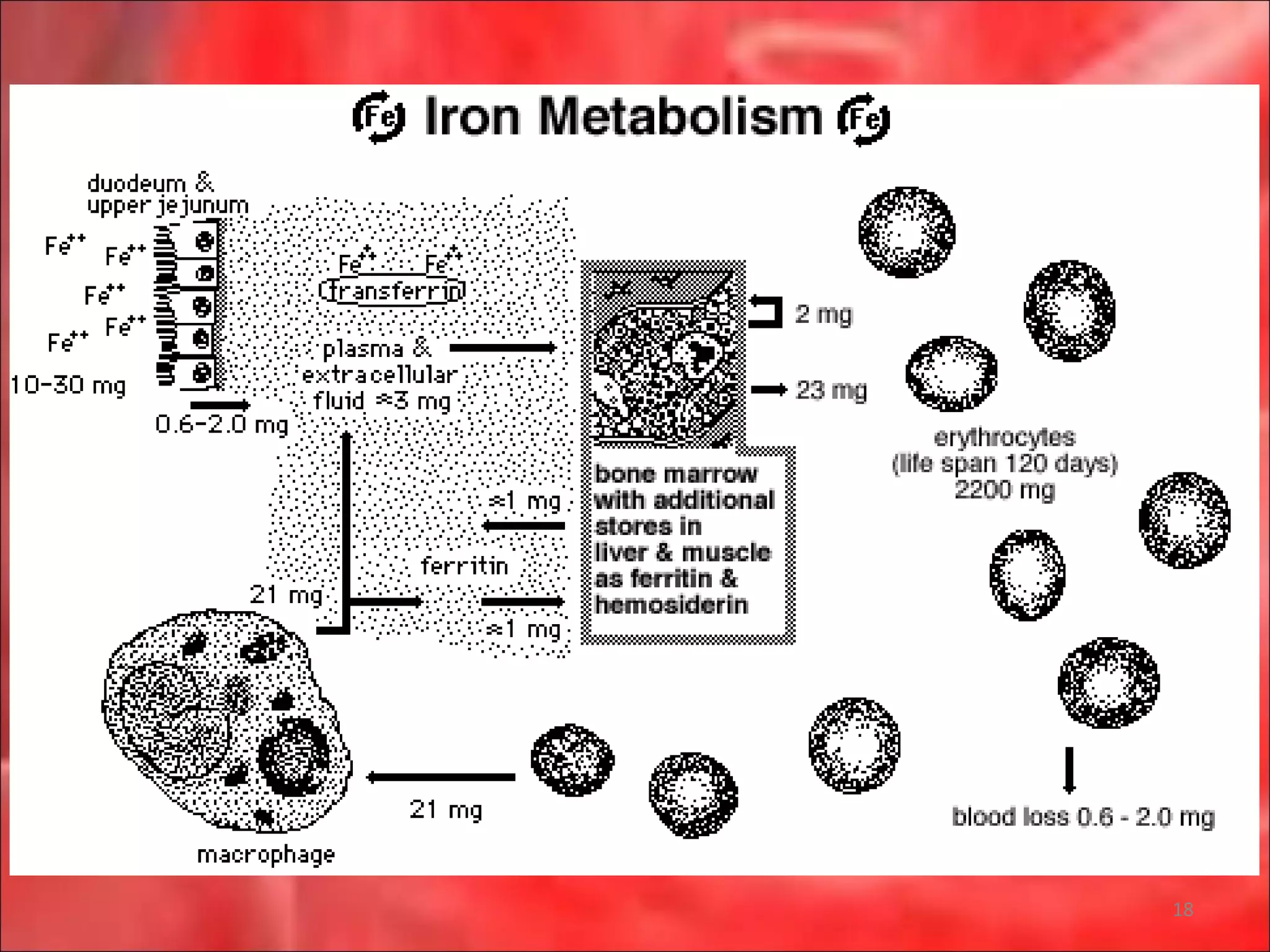
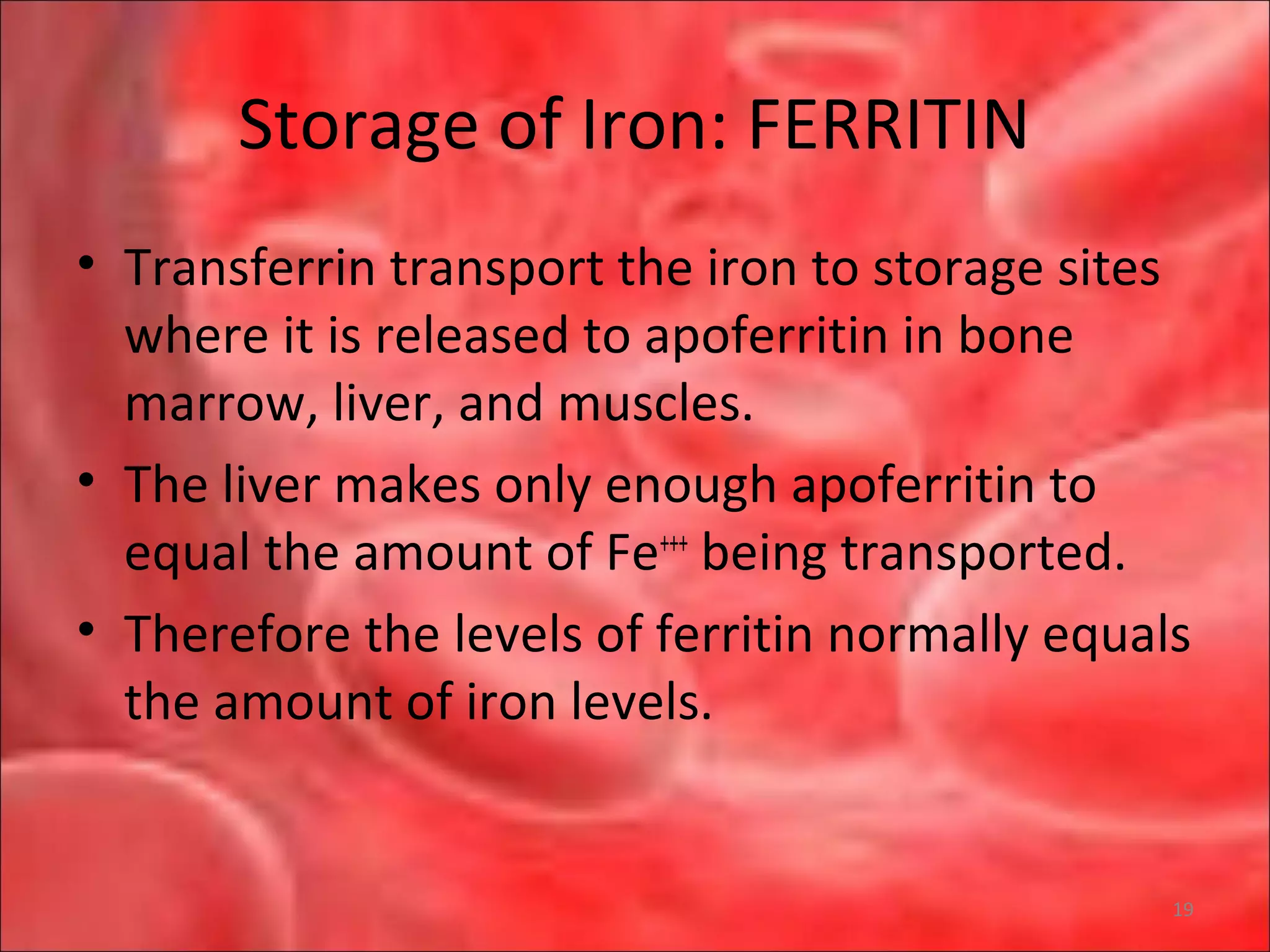
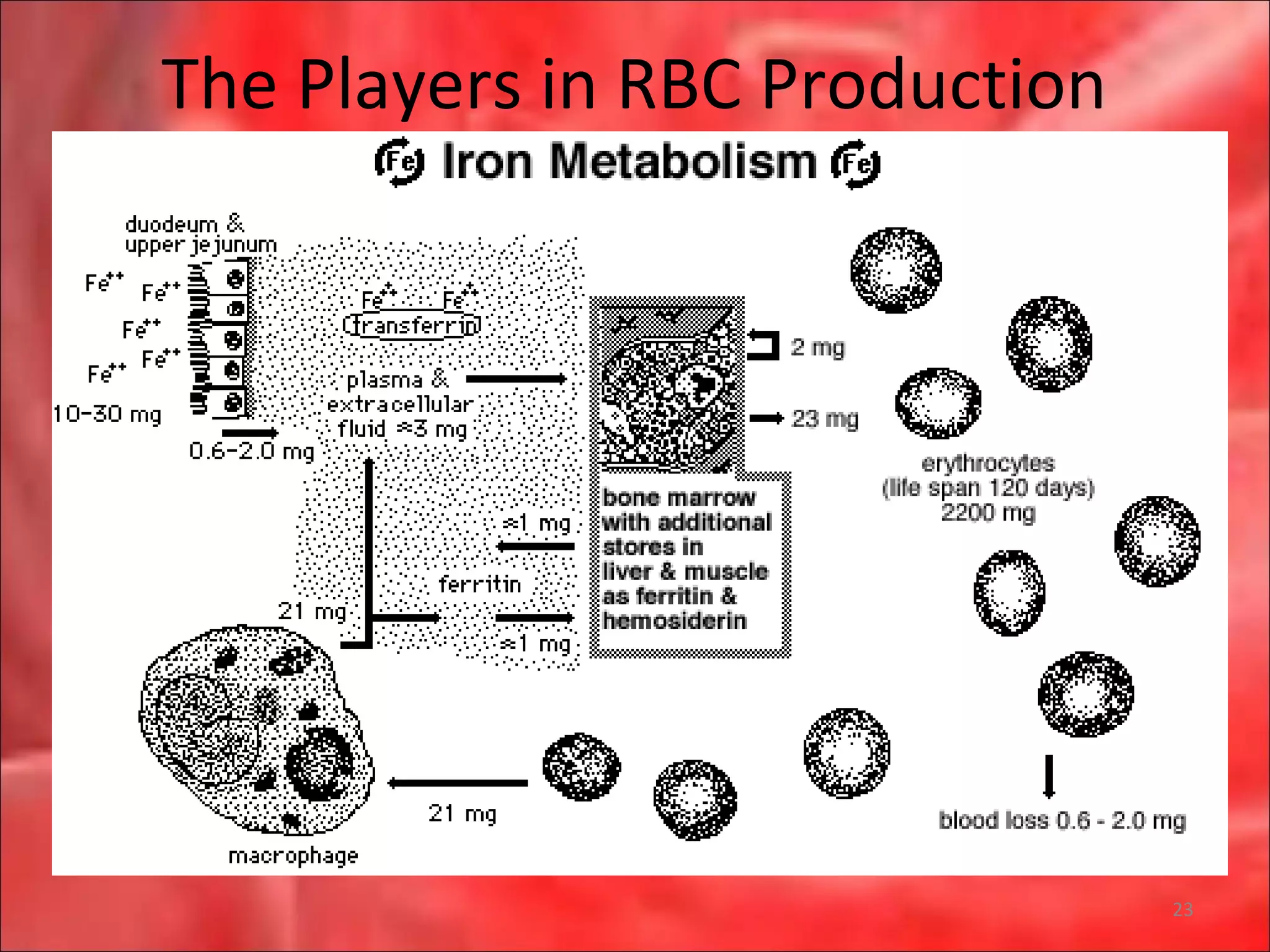
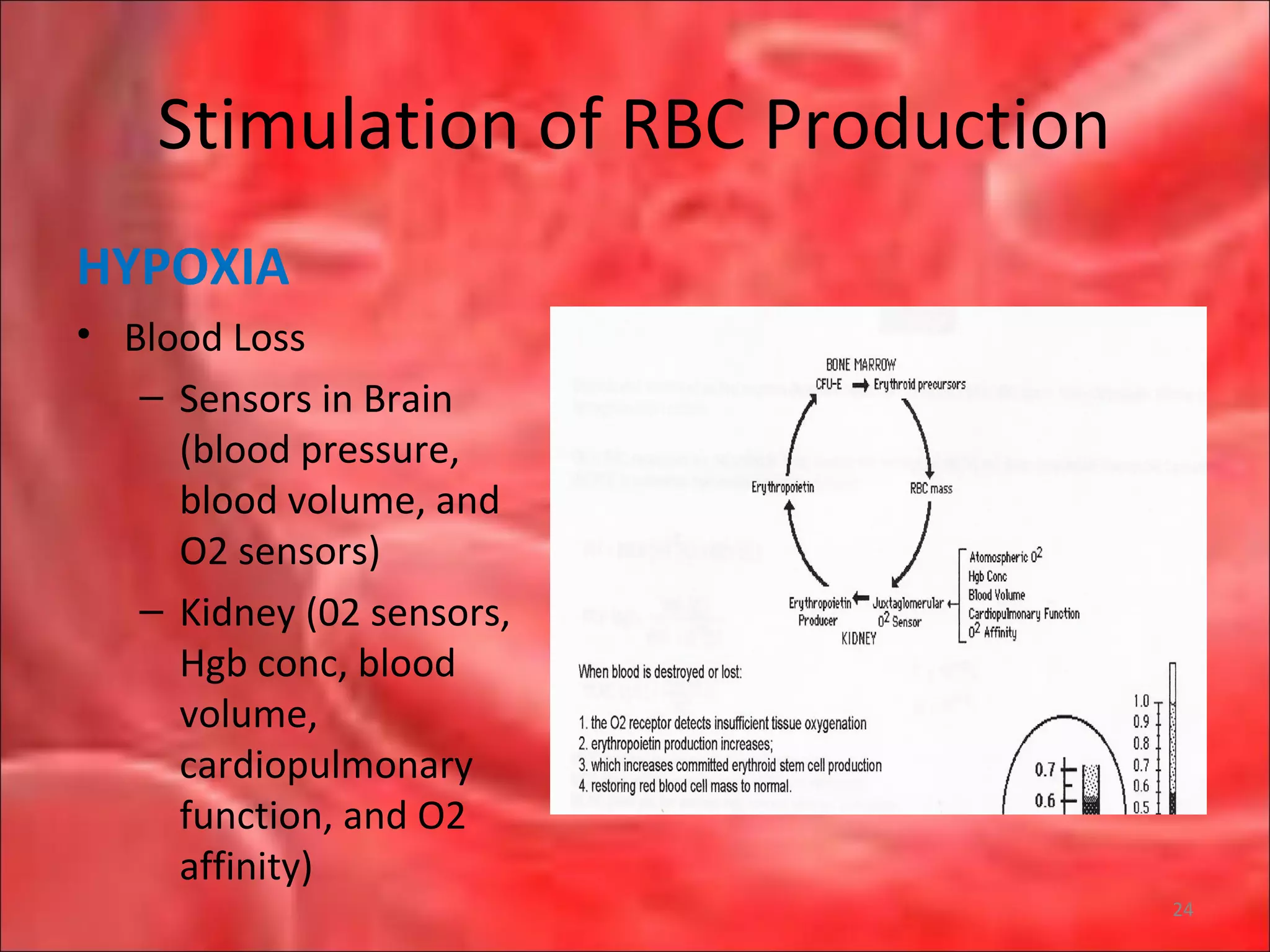
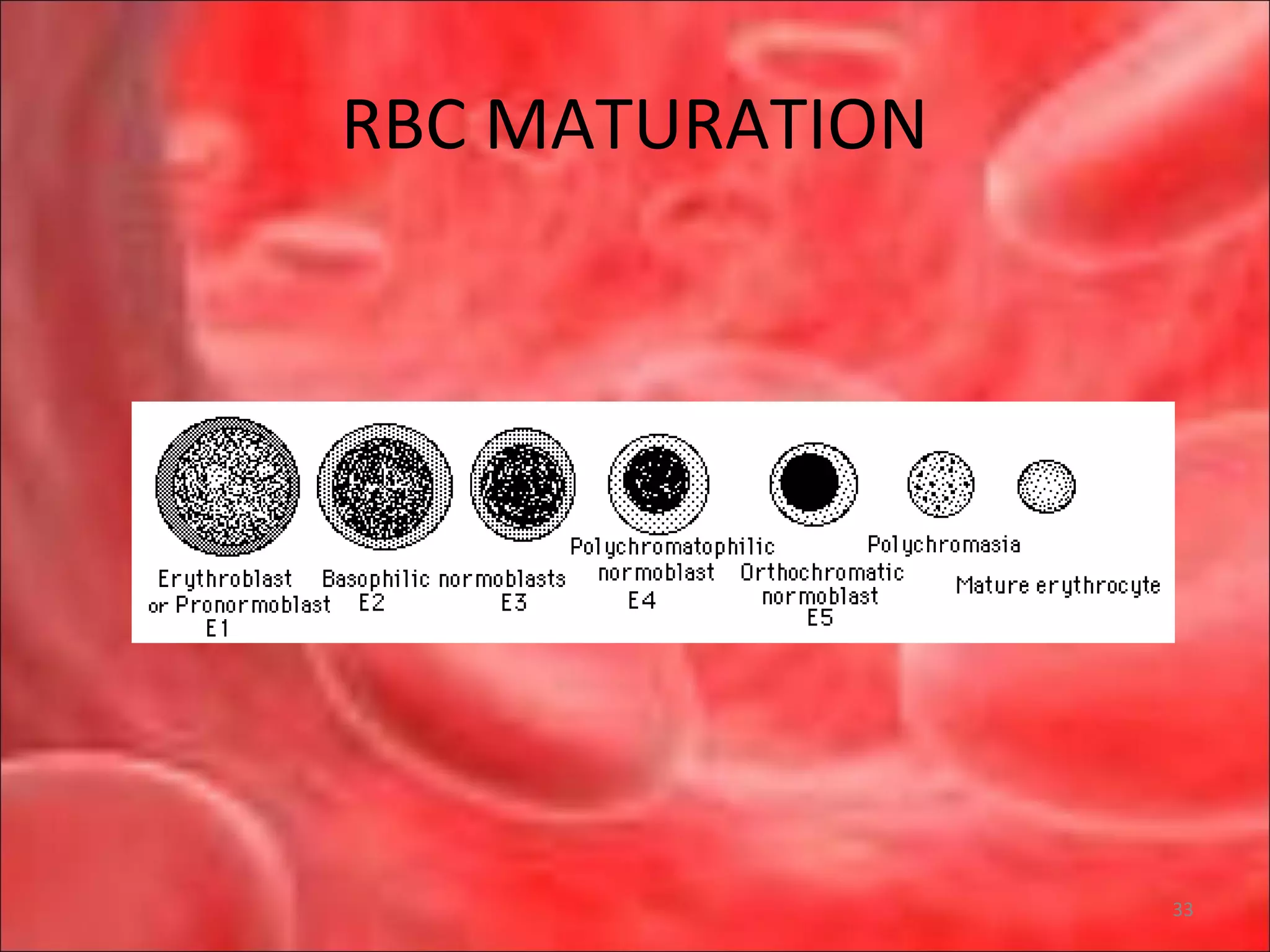
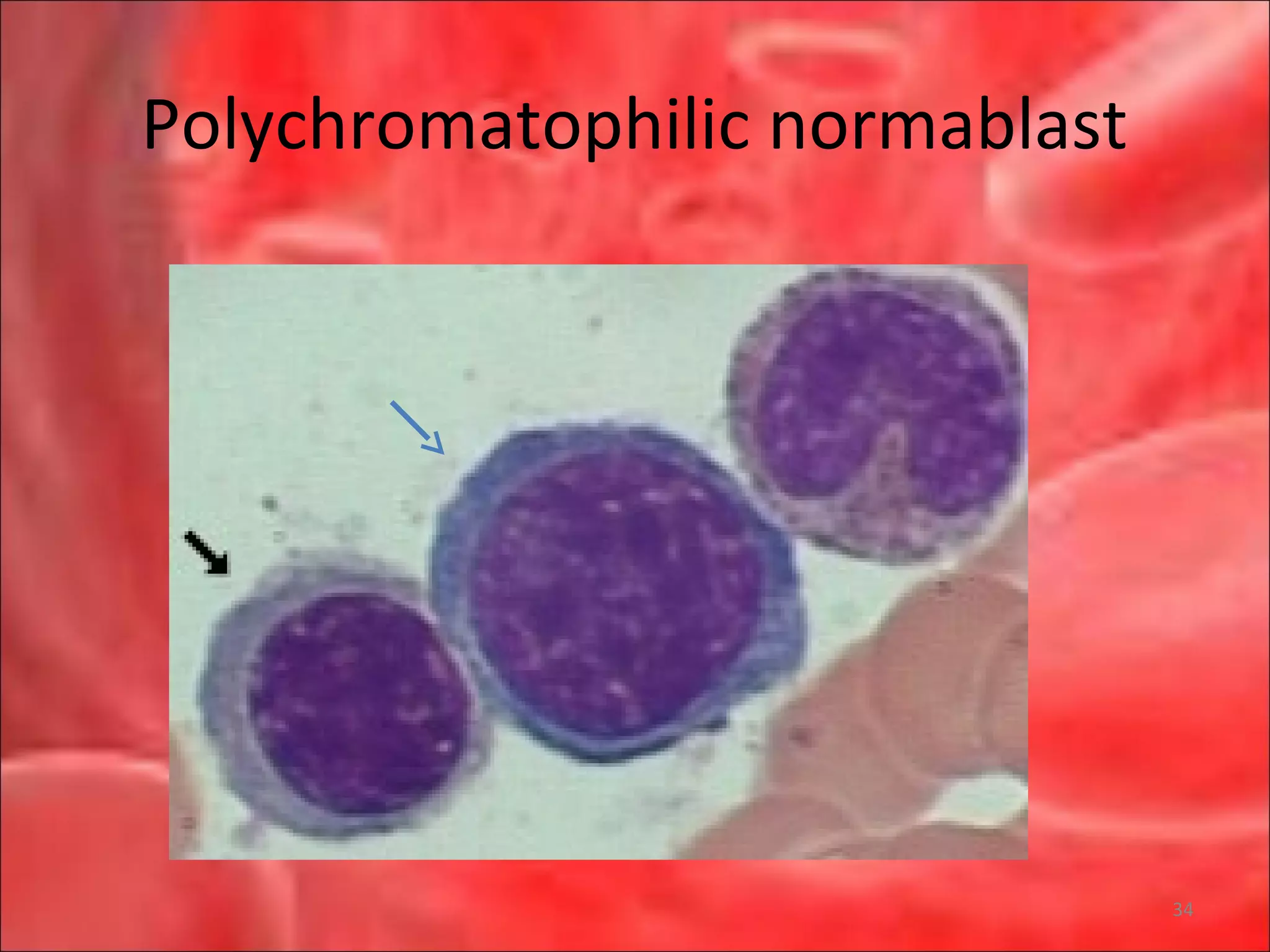
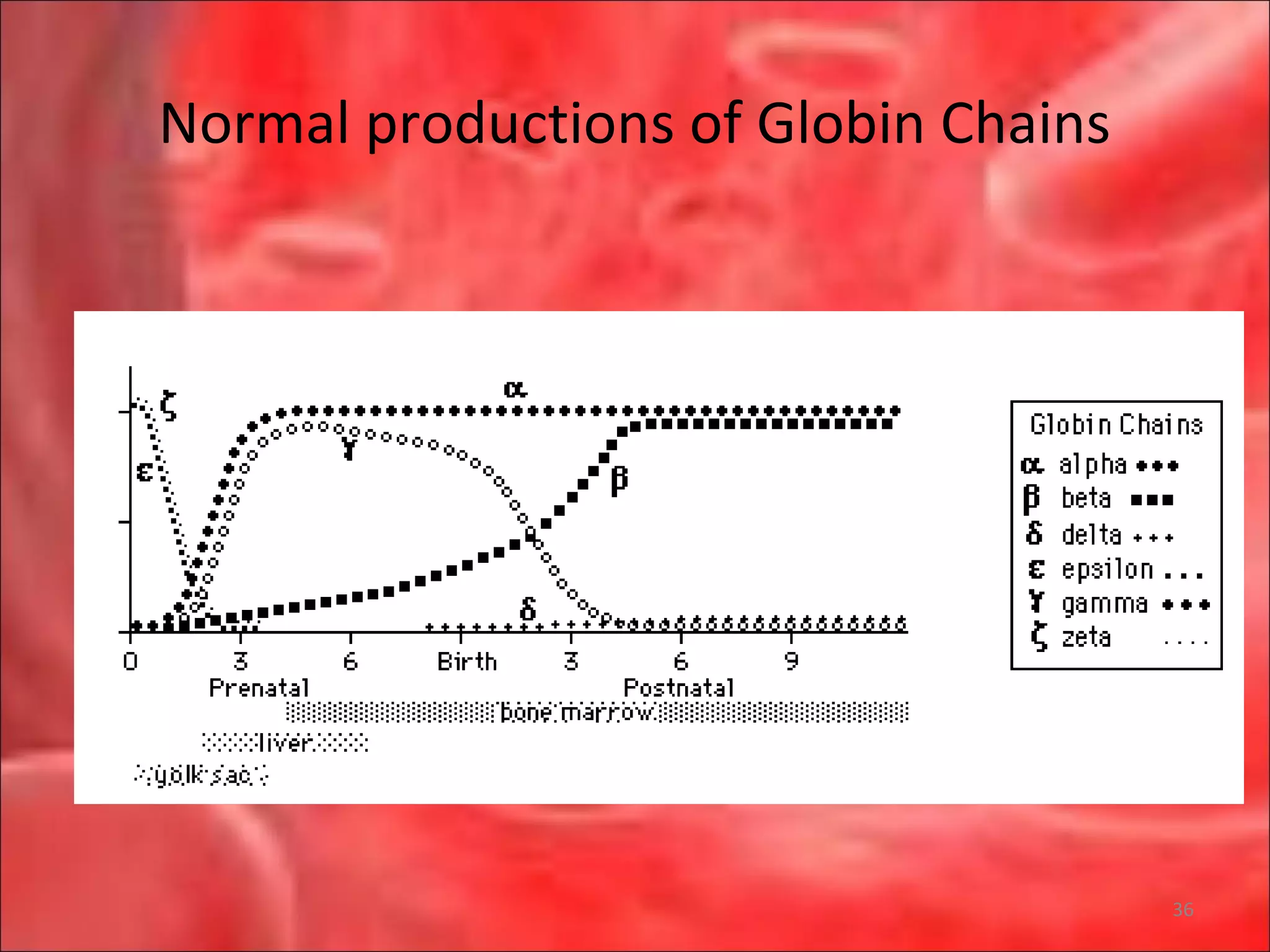
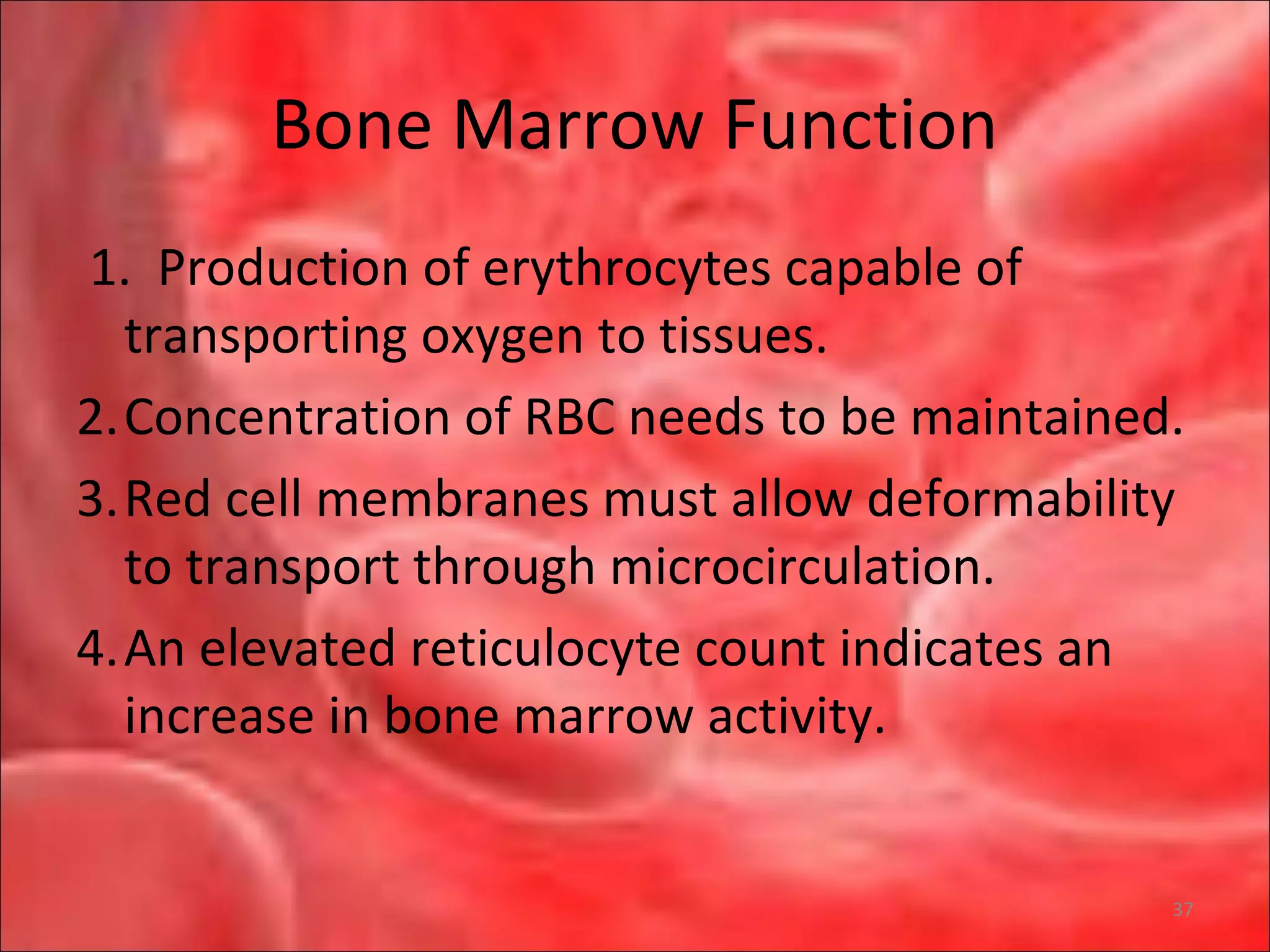
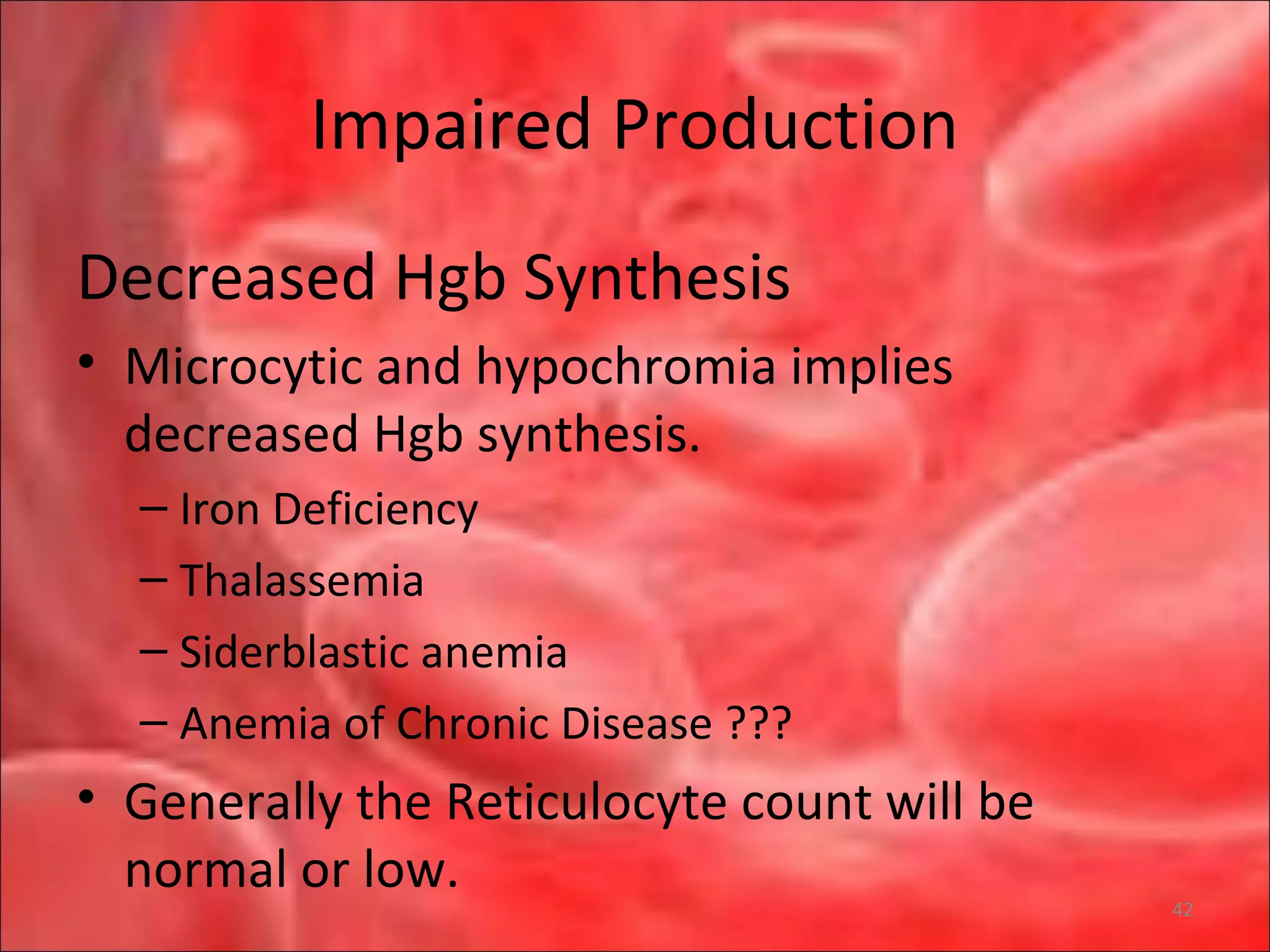
This document outlines the objectives and content of a lecture on anemias and red blood cell dyscrasias. The objectives cover understanding bone marrow regulation, causes of increased and decreased red blood cell production, hemolytic anemias, iron studies, and laboratory tests to diagnose specific anemias. The content discusses red blood cell development, iron metabolism, erythropoietin regulation, hemoglobin structure and types, and classifications of anemias including causes of impaired production and increased destruction.

























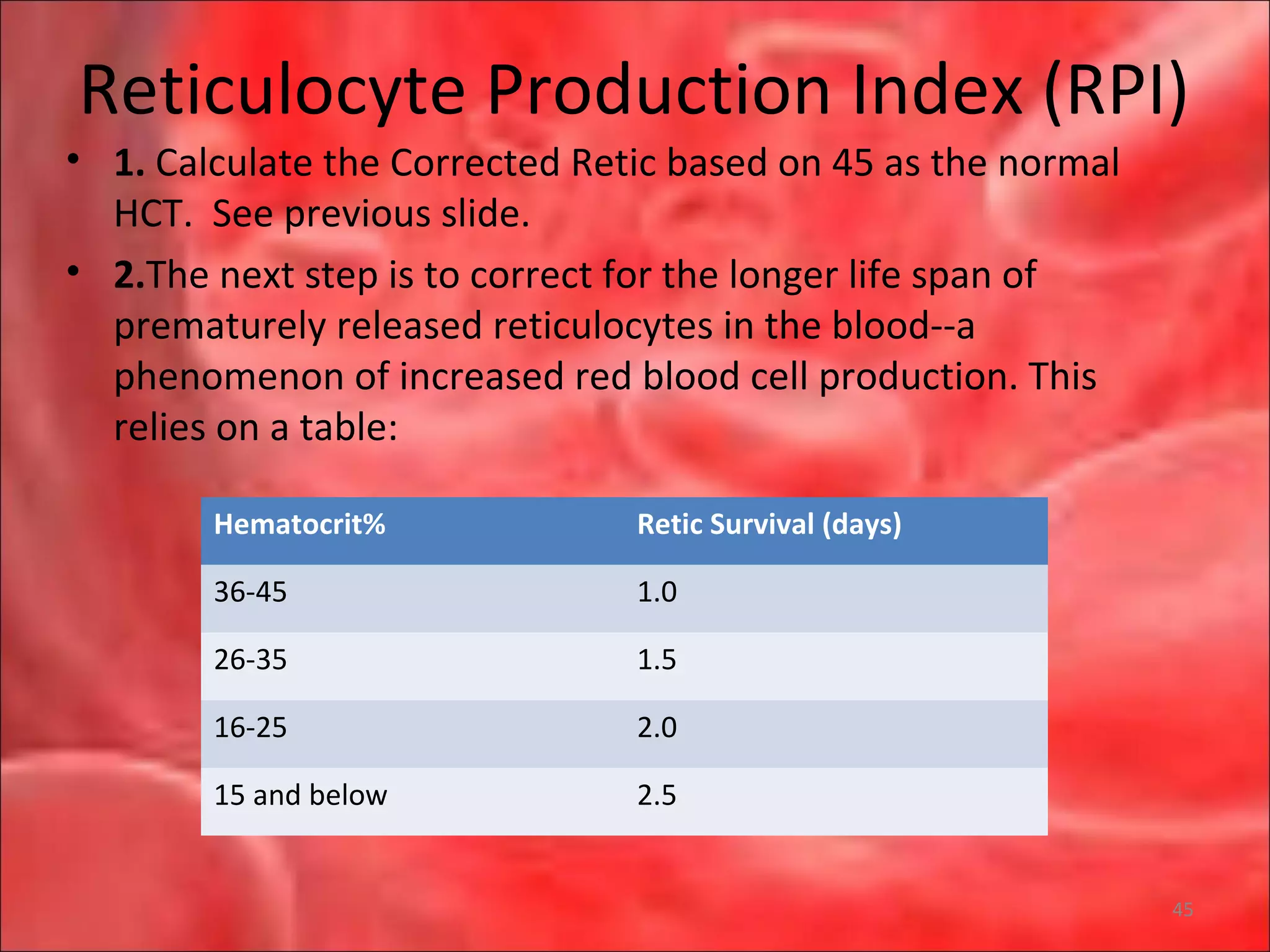
![RPI Calculation and Interpretation
• So, in a person whose uncorrected reticulocyte count is 5%,
hemoglobin 7.5 g/dL, hematocrit 25%, the RPI would be:
• [Corrrected retic]/[maturation correction] = [5 x (25/45)] /2 =
1.4
• Interpretation
• The reticulocyte index (RI) should be between 1.0 and 2.0 for
a healthy individual.
• RI < 2 with anemia indicates decreased production of
reticulocytes and therefore red blood cells.
• RI > 2 with anemia indicates loss of red blood cells
(destruction, bleeding, etc) leading to increased
compensatory production of reticulocytes to replace the lost
red blood cells. 46](https://image.slidesharecdn.com/anemias-interpretationofcbc2010rev-101123160852-phpapp01/75/Anemias-interpretation-of-cbc-2010rev-46-2048.jpg)