Downloaded 62 times













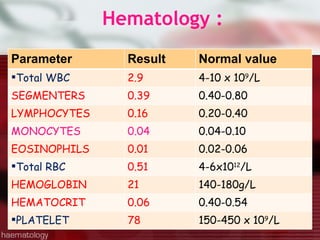
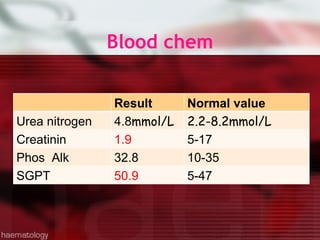



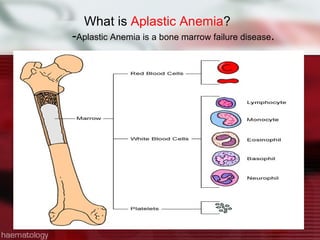

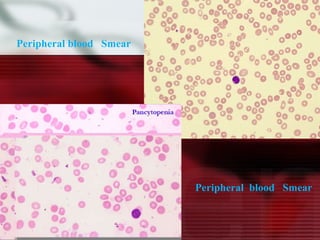
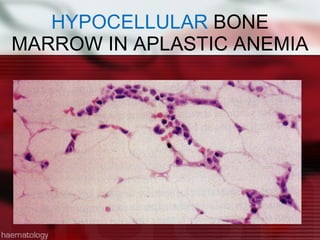




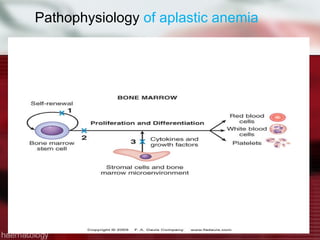






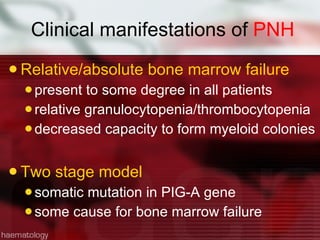
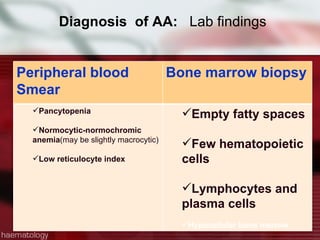

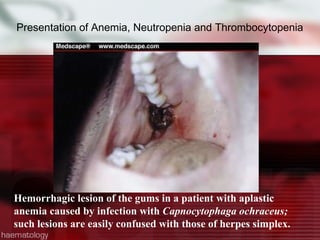
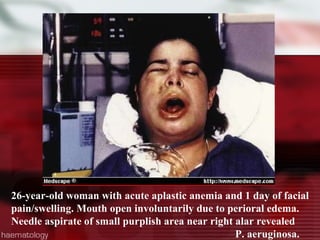
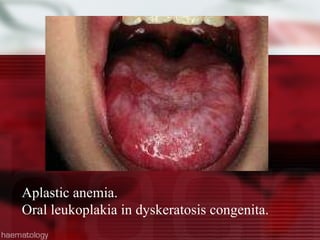







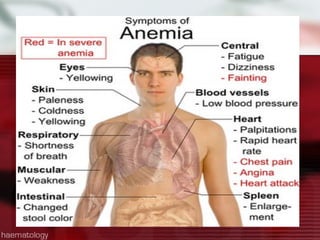


This patient is a 34-year-old Filipino man who presented with dizziness, loss of appetite, vomiting, and generalized weakness. His history and lab results are consistent with a diagnosis of aplastic anemia. Aplastic anemia is a bone marrow failure where the production of red blood cells, white blood cells, and platelets is suppressed, leading to pancytopenia. Treatment options include supportive care like transfusions, immunosuppressive drugs, and bone marrow transplant, with outcomes varying based on a patient's age and availability of a histocompatible donor.



































































