Downloaded 501 times










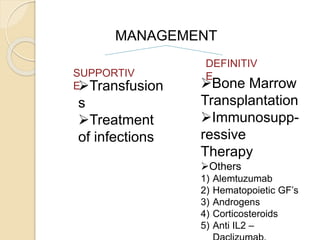

















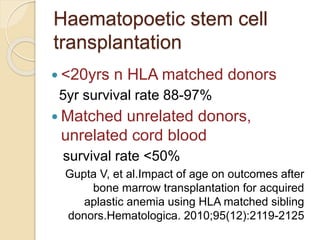



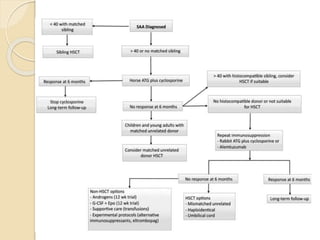


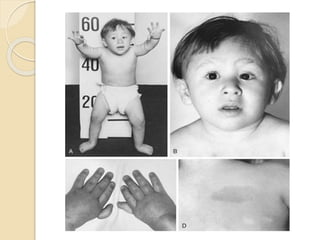





This document summarizes the management of aplastic anemia. It discusses the classification, pathogenesis, clinical evaluation, investigations and definitive management including immunosuppressive therapy with antithymocyte globulin and cyclosporine. It also covers supportive management with transfusions, antibiotics and growth factors. Treatment failure options and hematopoietic stem cell transplantation are described. Inherited causes like Fanconi anemia are also summarized.



































































