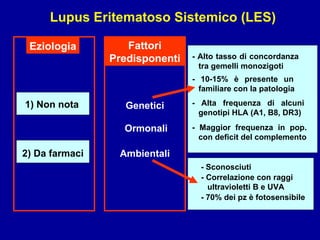
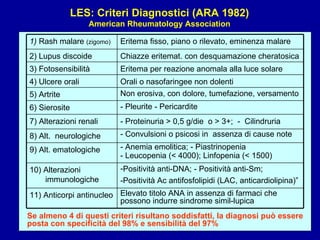
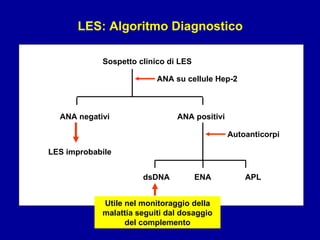
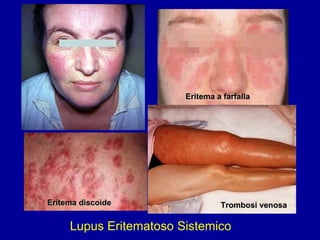
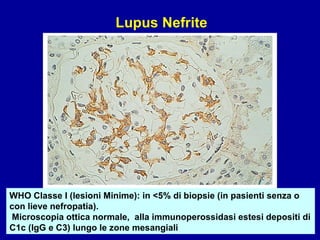
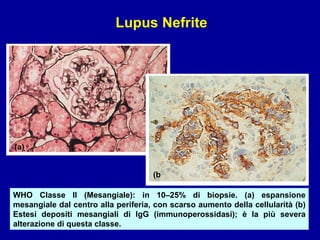
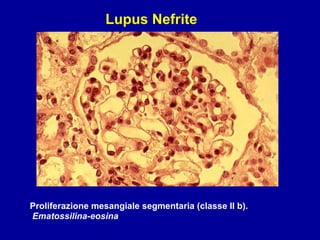
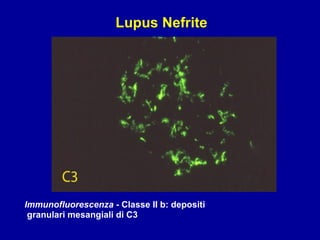
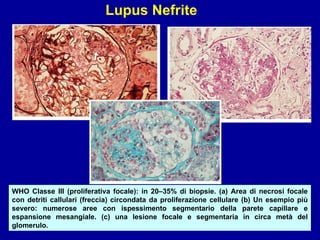
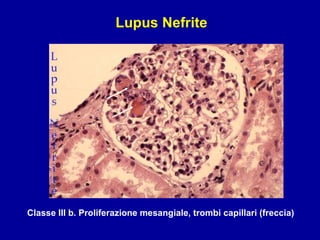
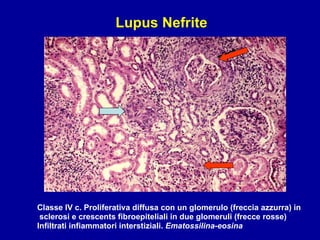
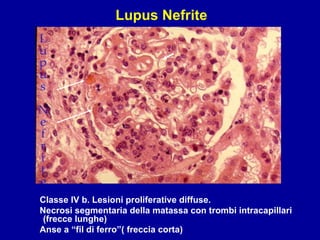
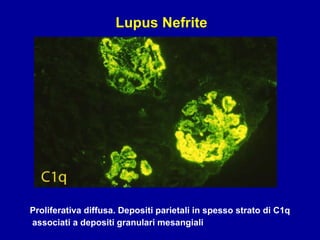
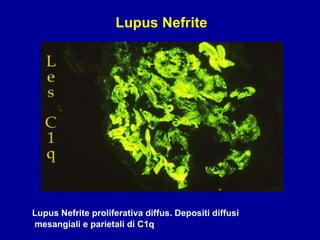
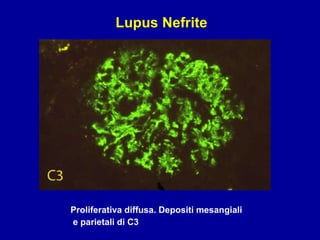
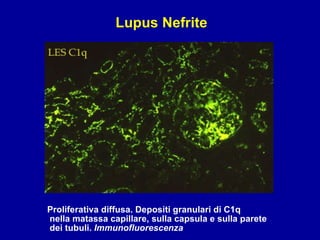
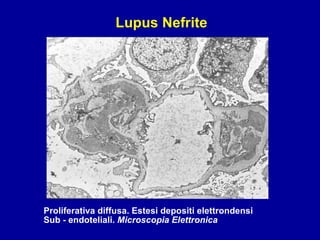
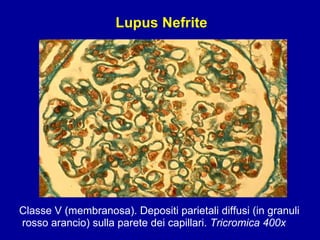
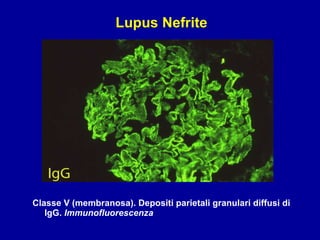
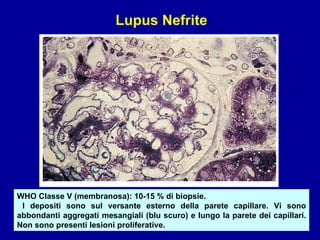
Il documento discute le mancani nefropatie e il lupus eritematoso sistemico (LES), descrivendo eziologia, patogenesi e criteri diagnostici. Esamina le manifestazioni cliniche e laboratoristiche della malattia, nonché le complicanze renali, in particolare la nefrite lupica. Infine, fornisce informazioni sulla classificazione della nefrite e le sue implicazioni terapeutiche.