Downloaded 26 times



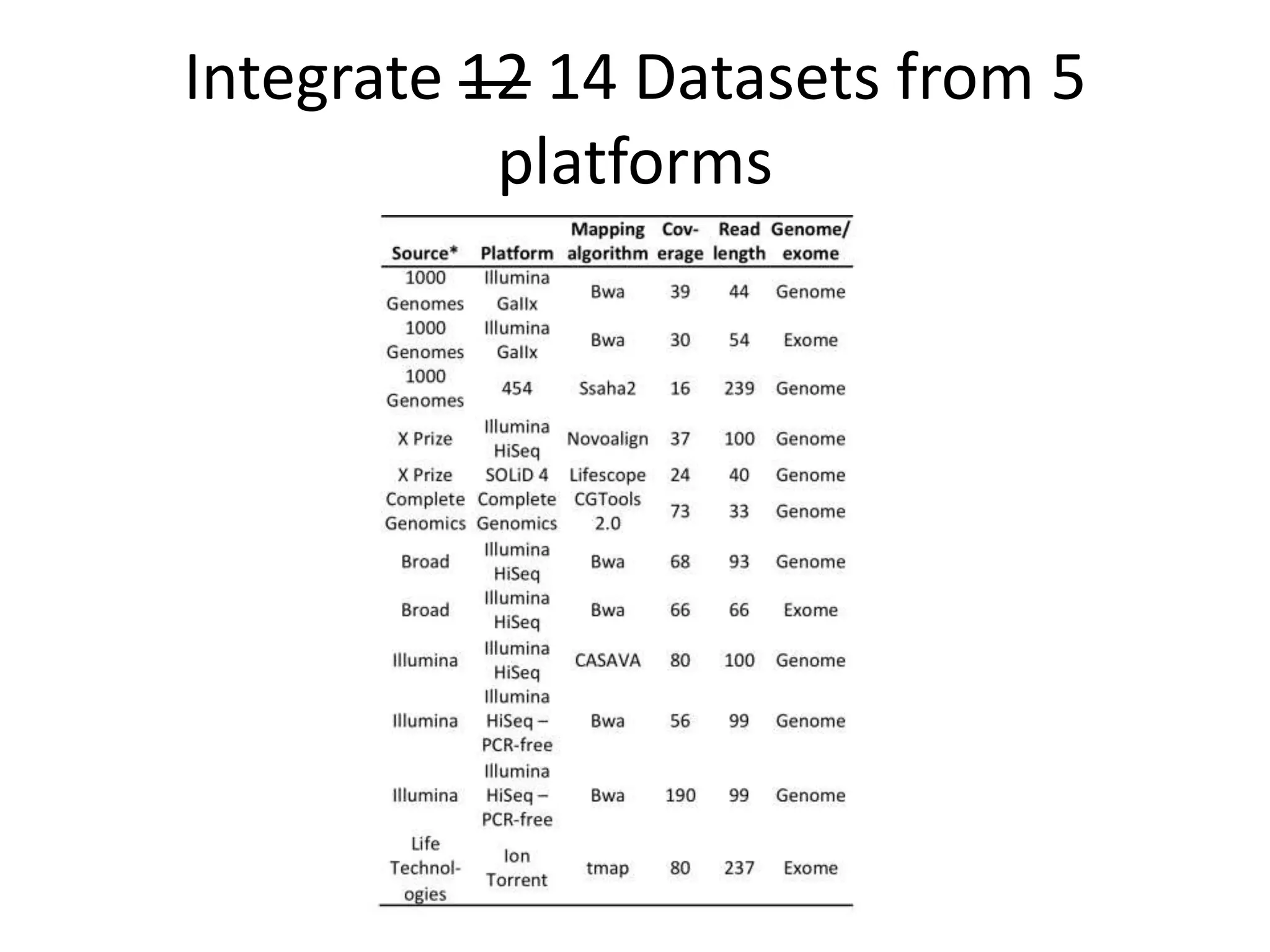


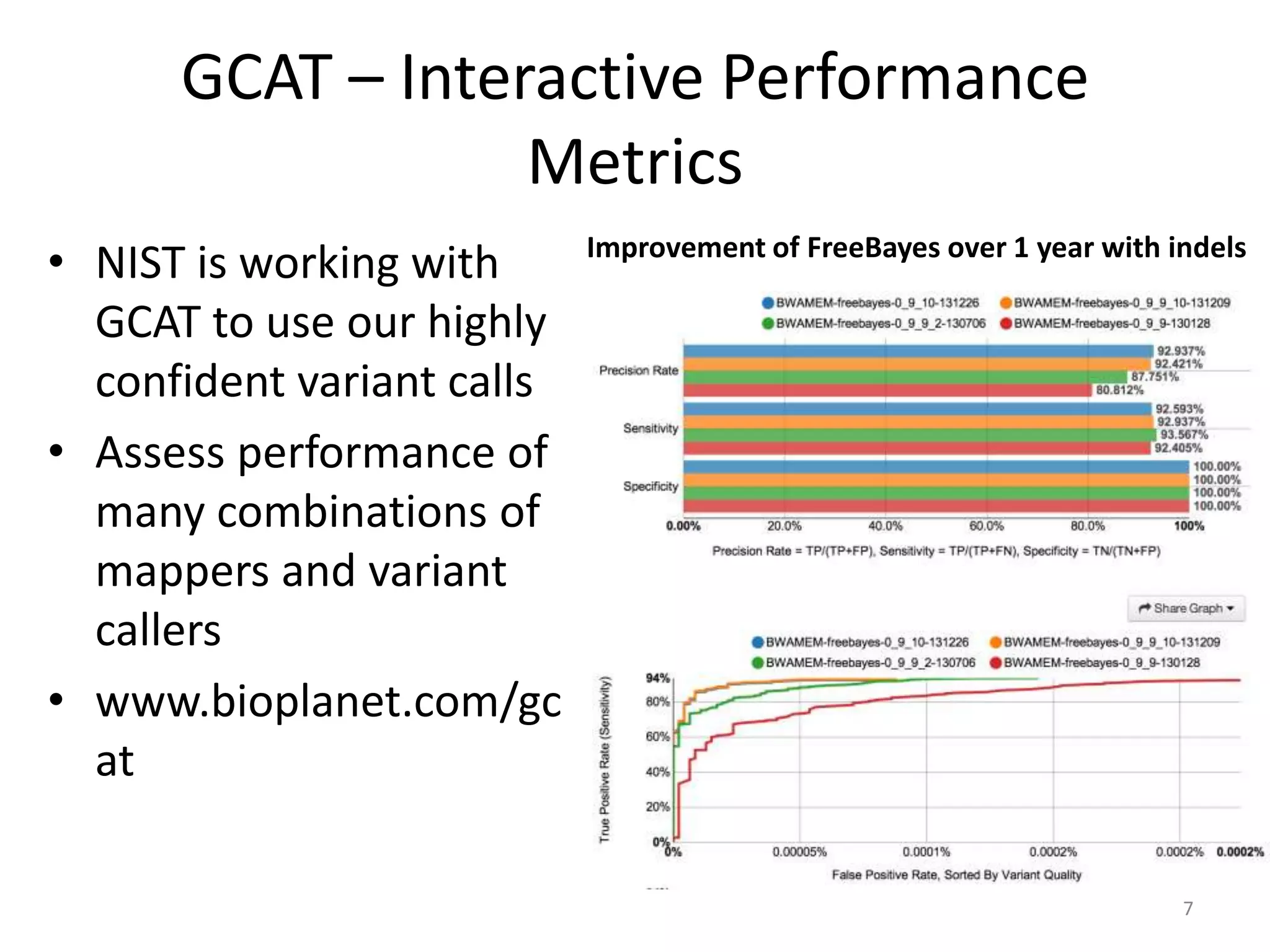

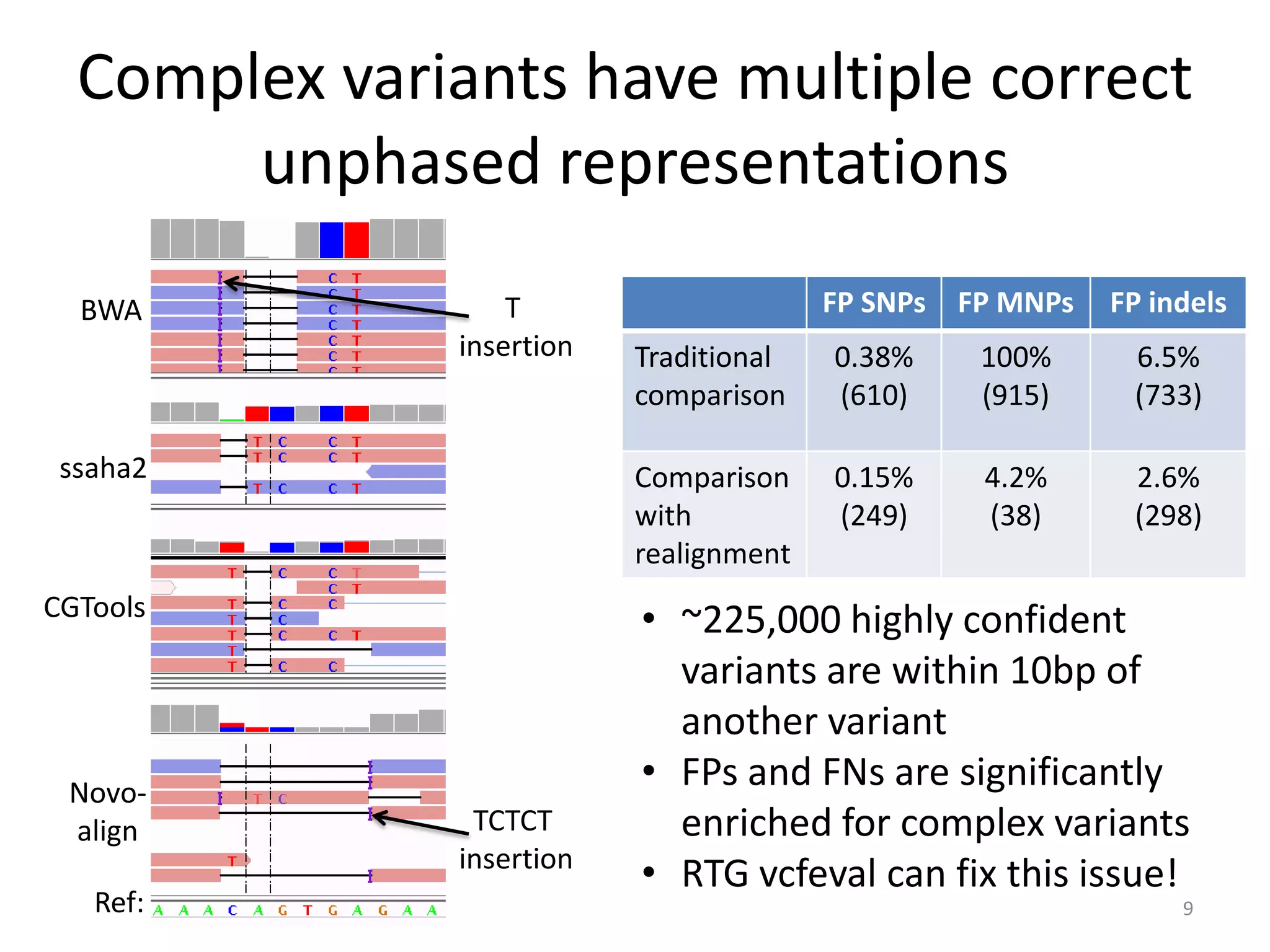
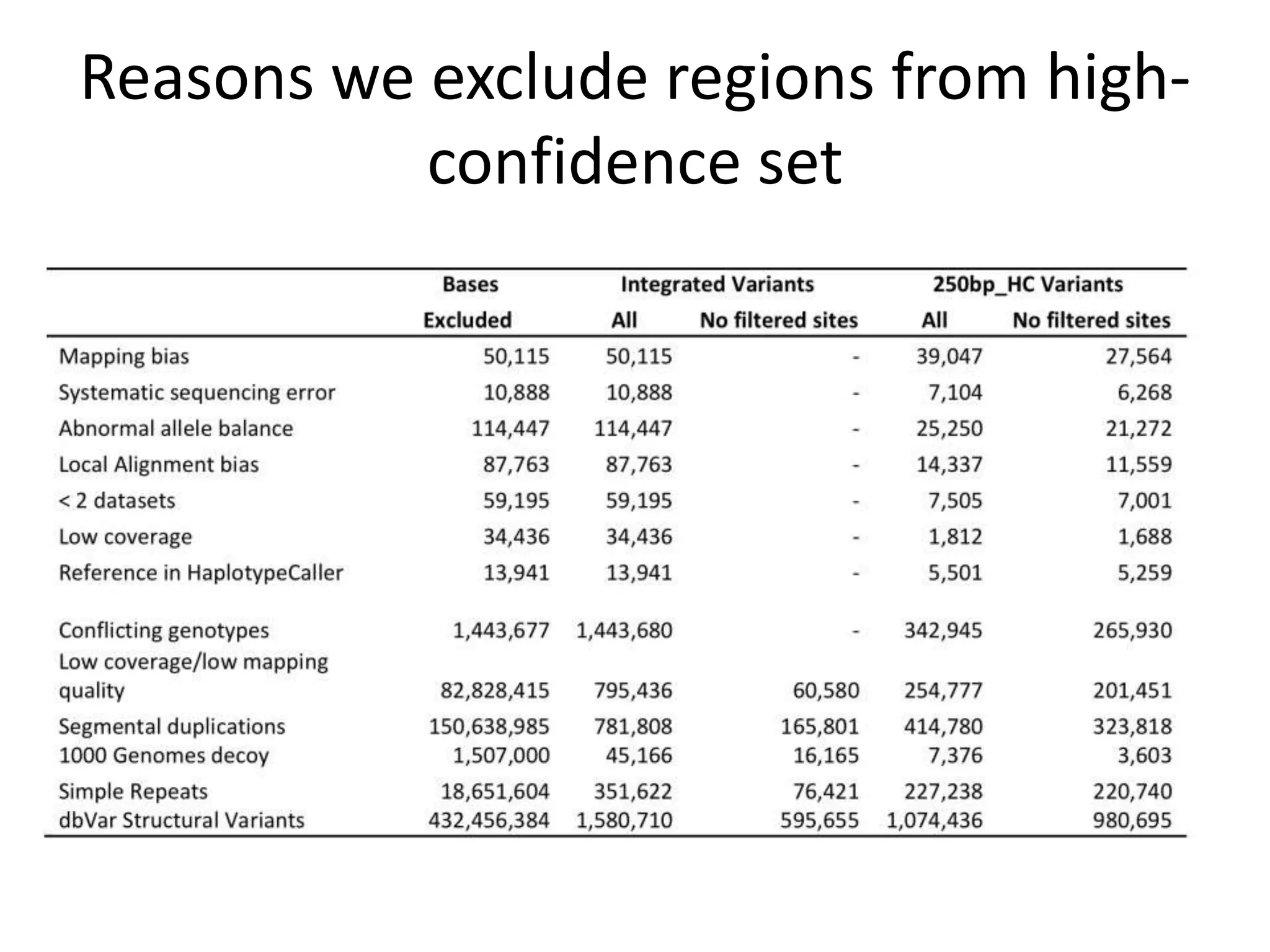
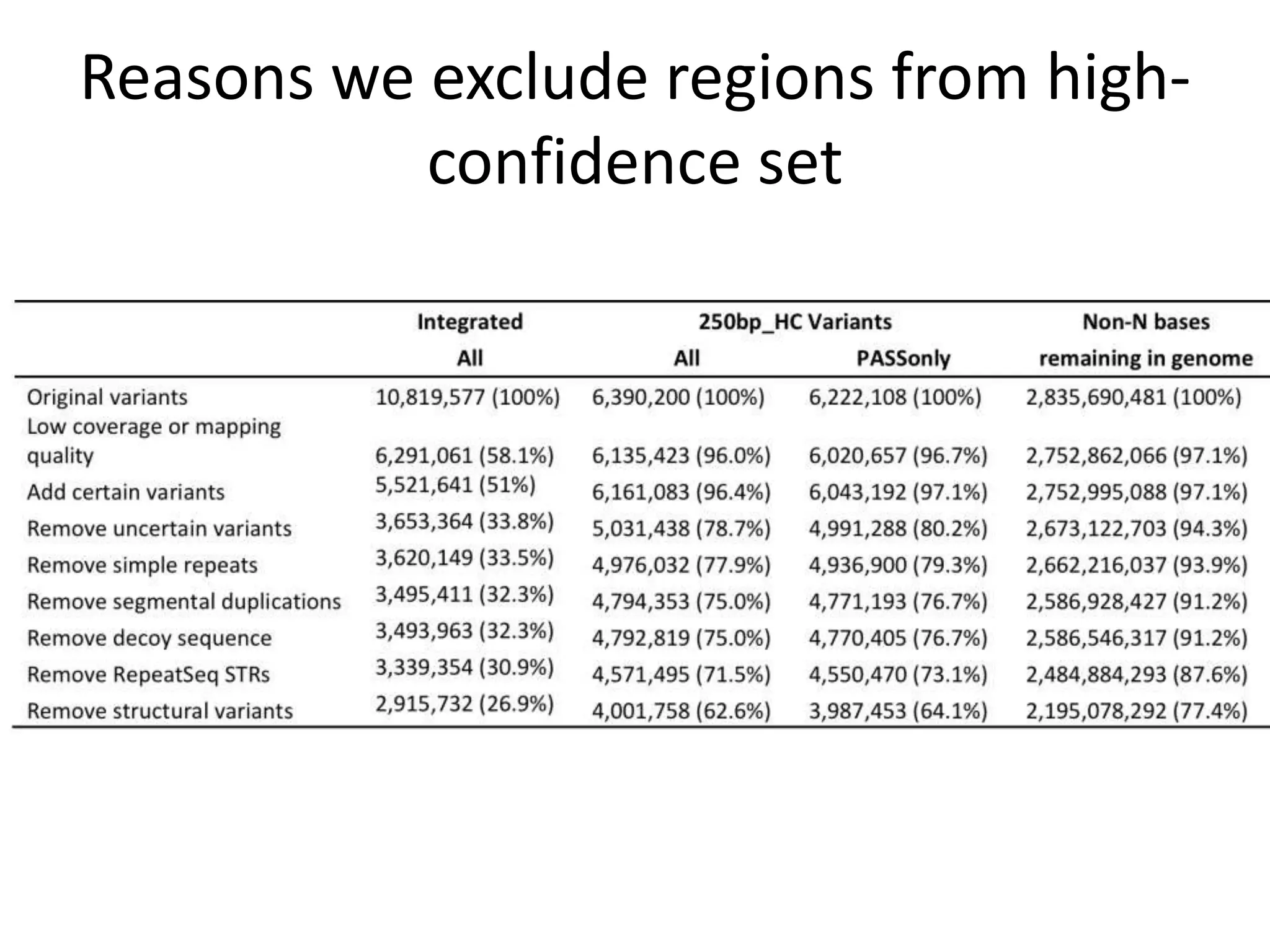
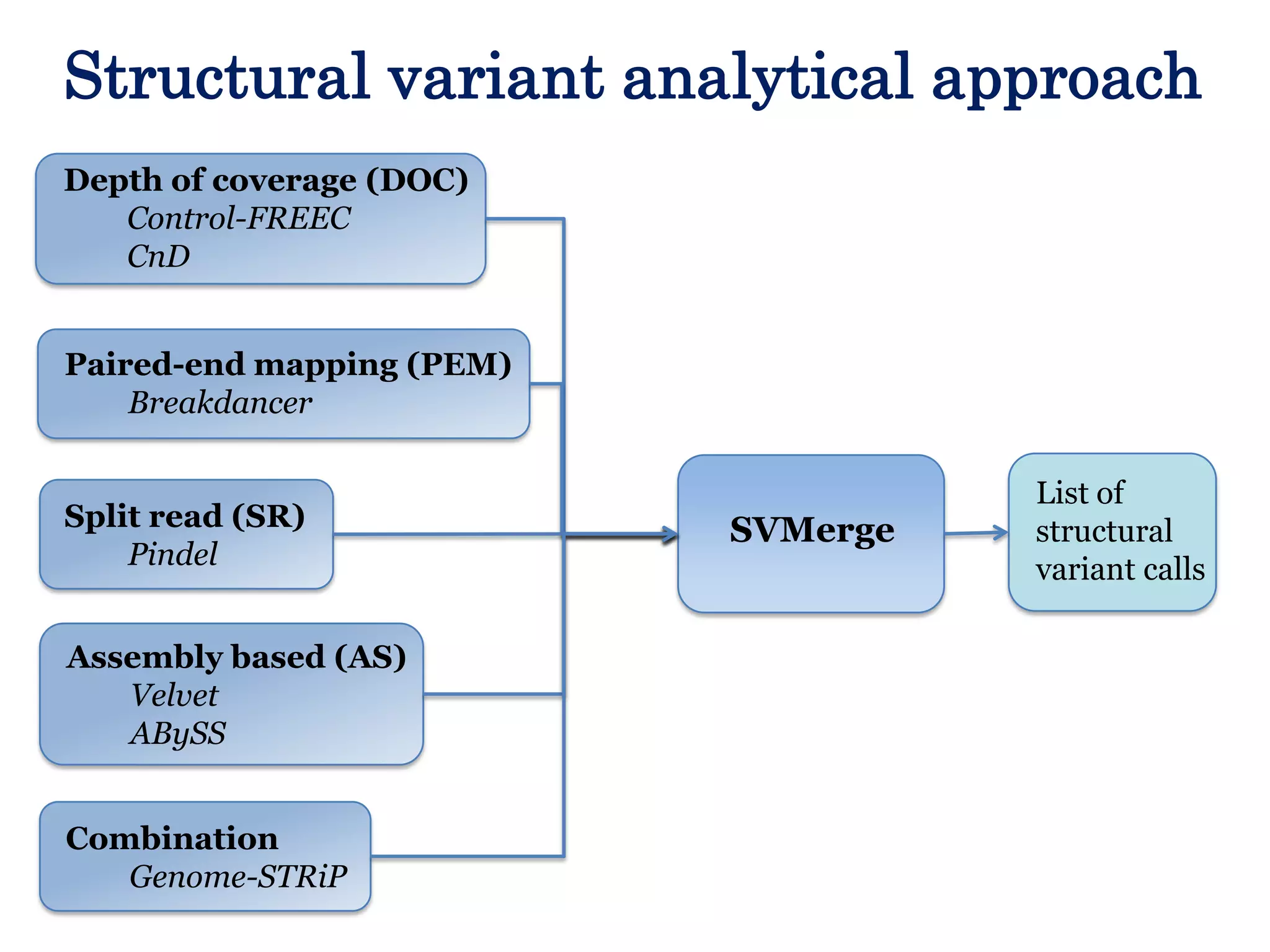
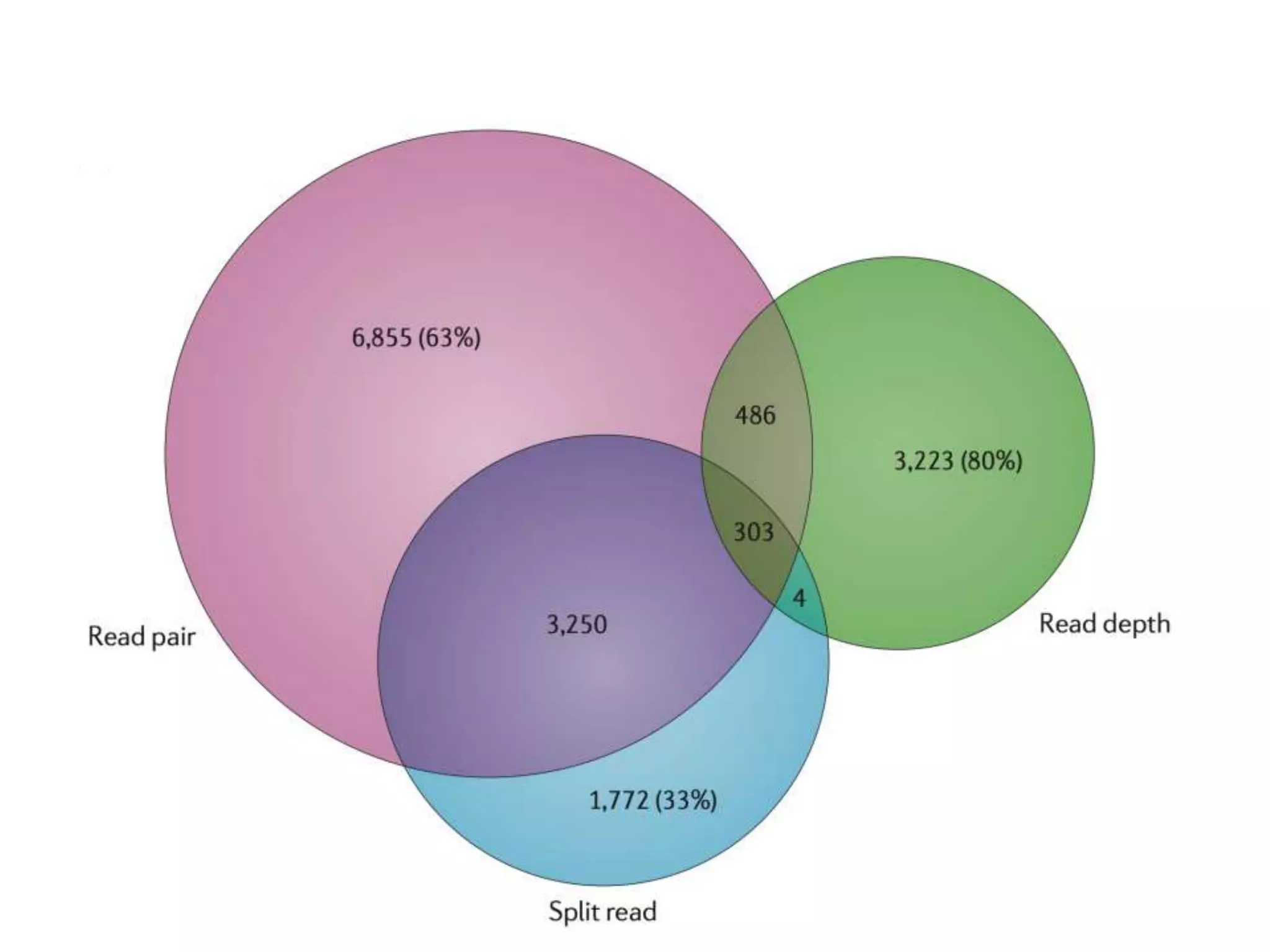


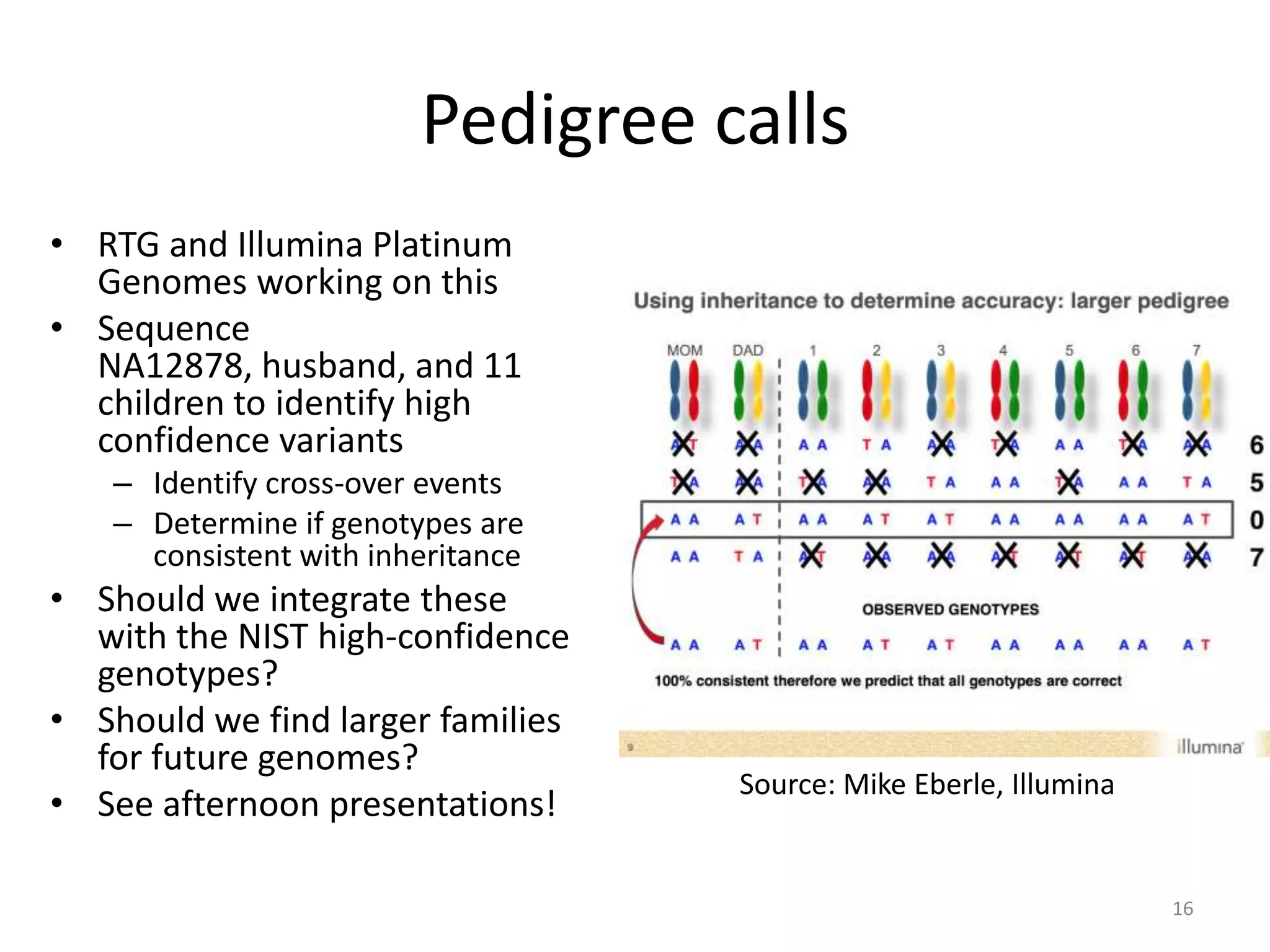
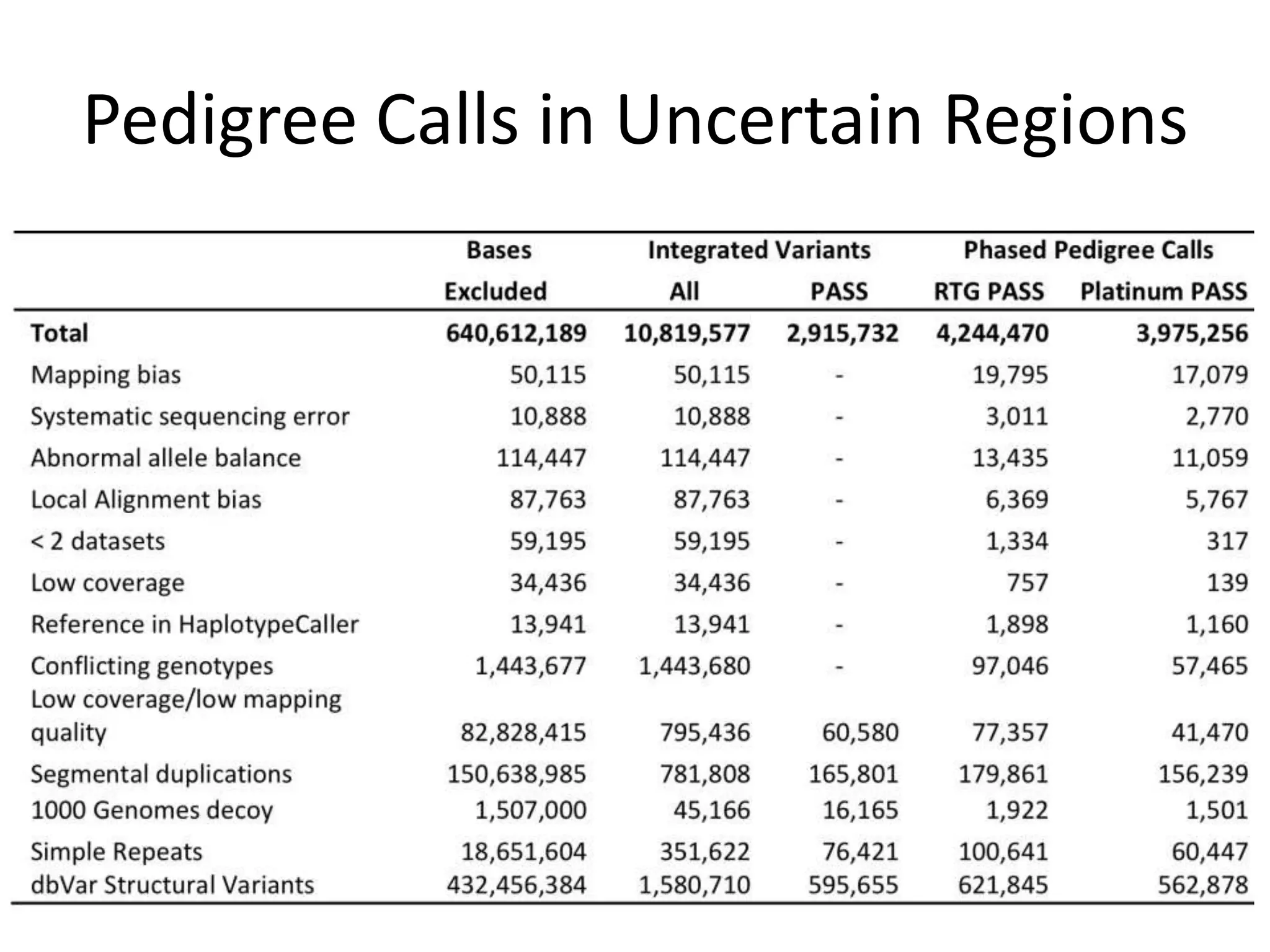










The Genome in a Bottle Consortium provided a progress update on integrating datasets from multiple sequencing platforms to generate highly confident genotype calls for use as a reference material. They integrated 12 datasets from 5 platforms and used evidence of bias and concordance across datasets to identify confident regions. They are working to characterize more complex and uncertain regions, and to validate calls using pedigree and long read sequencing data. Future work includes characterizing additional genomes from diverse populations and cancer samples.























































































