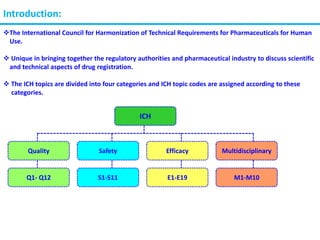
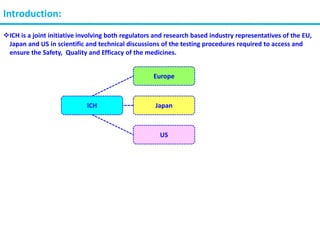
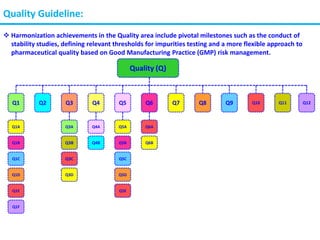
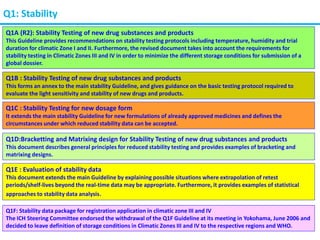
The document summarizes the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) quality guidelines. The ICH brings together regulatory authorities and the pharmaceutical industry to discuss drug registration. The quality guidelines cover topics such as stability testing, validation of analytical procedures, impurities, pharmacopoeias, quality of biotechnological products, specifications, good manufacturing practices, pharmaceutical development, quality risk management, and the pharmaceutical quality system.


















































![ICH [ Q ] Guidelines](https://cdn.slidesharecdn.com/ss_thumbnails/ichabhishek-210812054107-thumbnail.jpg?width=640&height=640&fit=bounds)


























