


















![Drug Master file Process
Two copies of the Drug Master File with one signed original of the covering letter and other necessary documents are send to the FDA’s
Central Drug Evaluation and Research (CDRL). [1]
The Drug Master File staff will audit the non-technical information for completeness and adequacy for submission. If the key elements are
missing, the staff will contact the proposed holder to try to obtain the necessary documents in order to file the DMF.
Once the DMFs are determined to be acceptable for filing, the document room staff assigns a DMF number and a letter is sent to the
contact person listed in the DMF.
All DMF submitted in CTD format is accepted by any authorities.](https://image.slidesharecdn.com/dmfanthonycrasto-141024000842-conversion-gate02/75/DMF-by-Anthony-Crasto-20-2048.jpg)








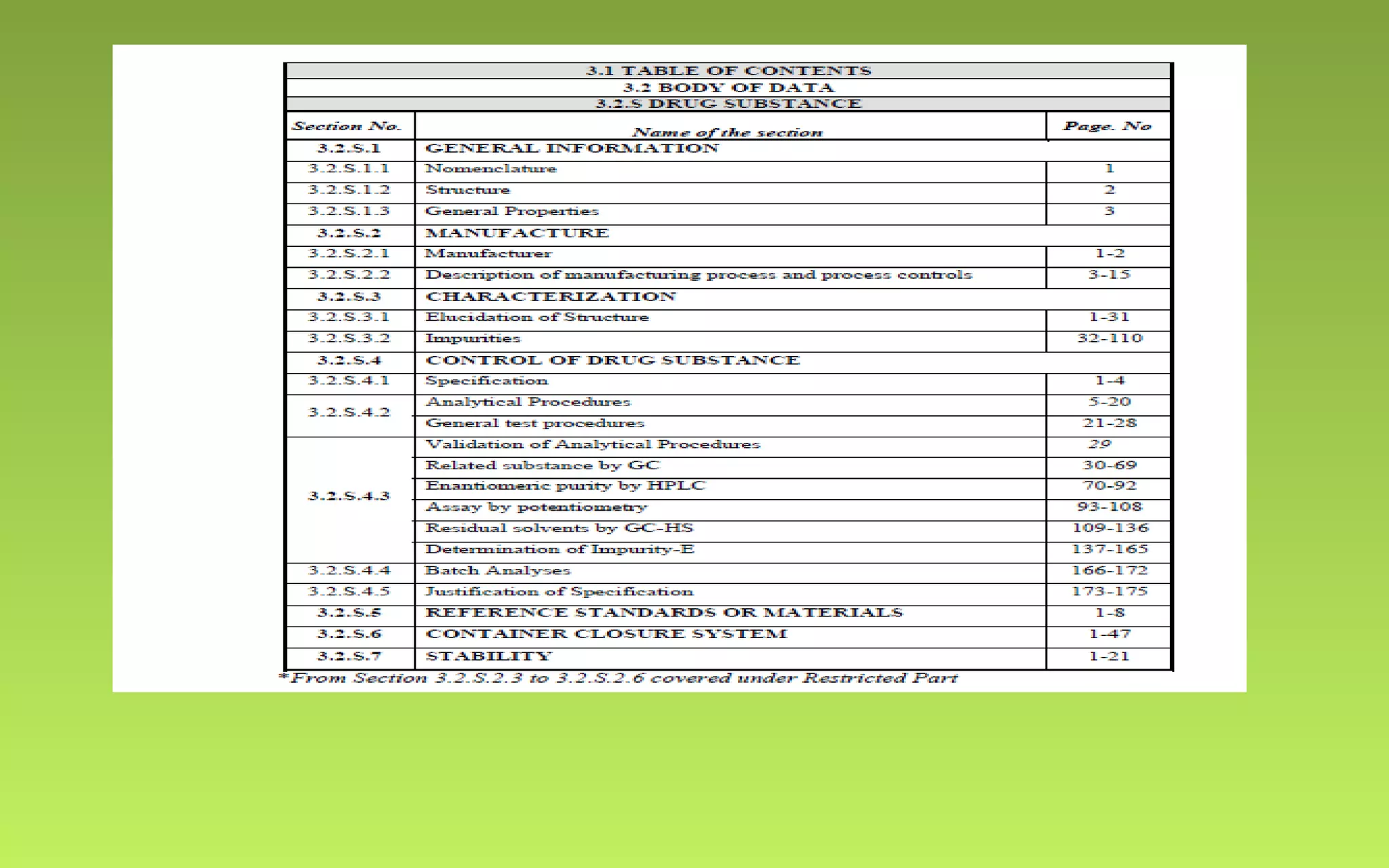
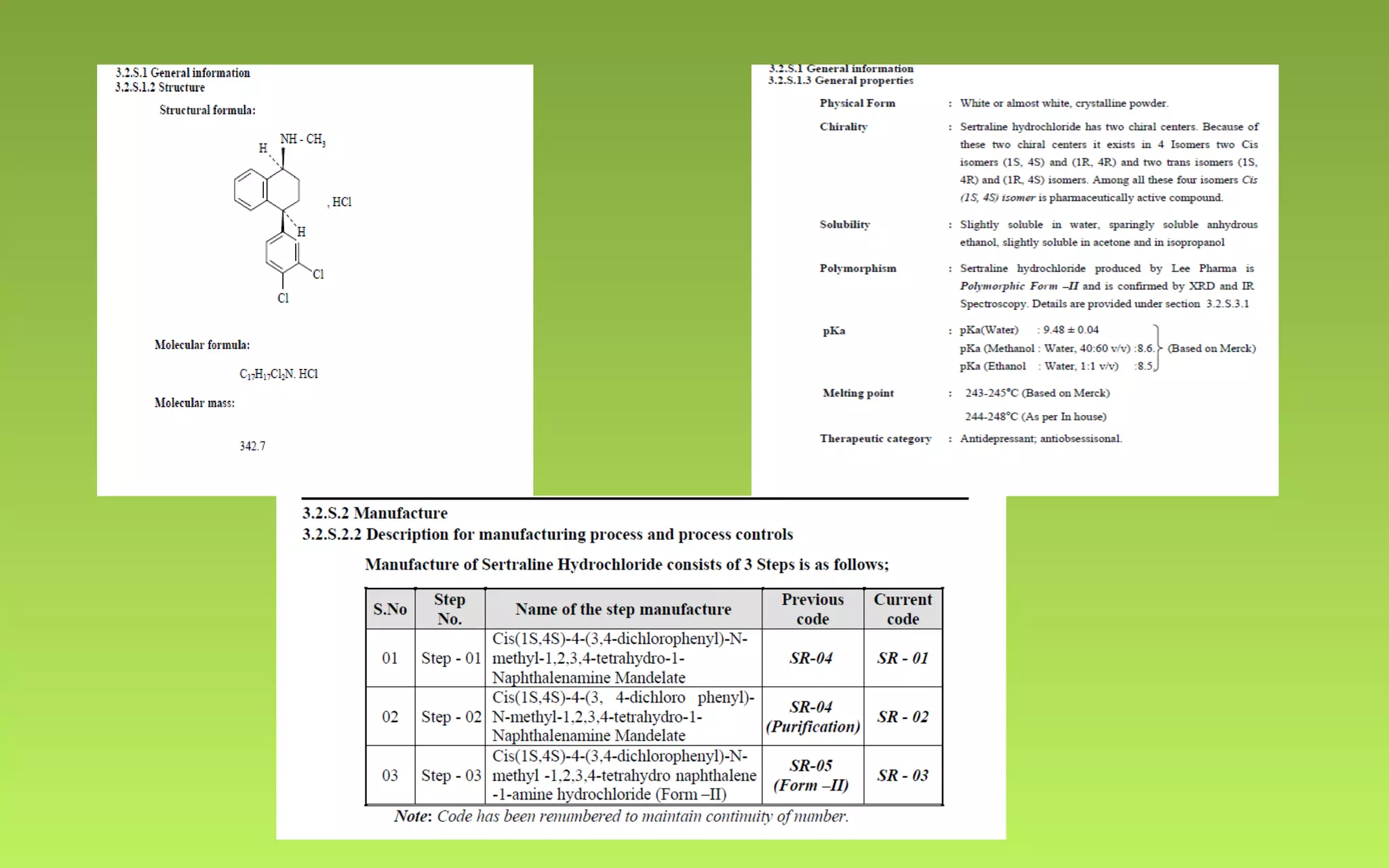

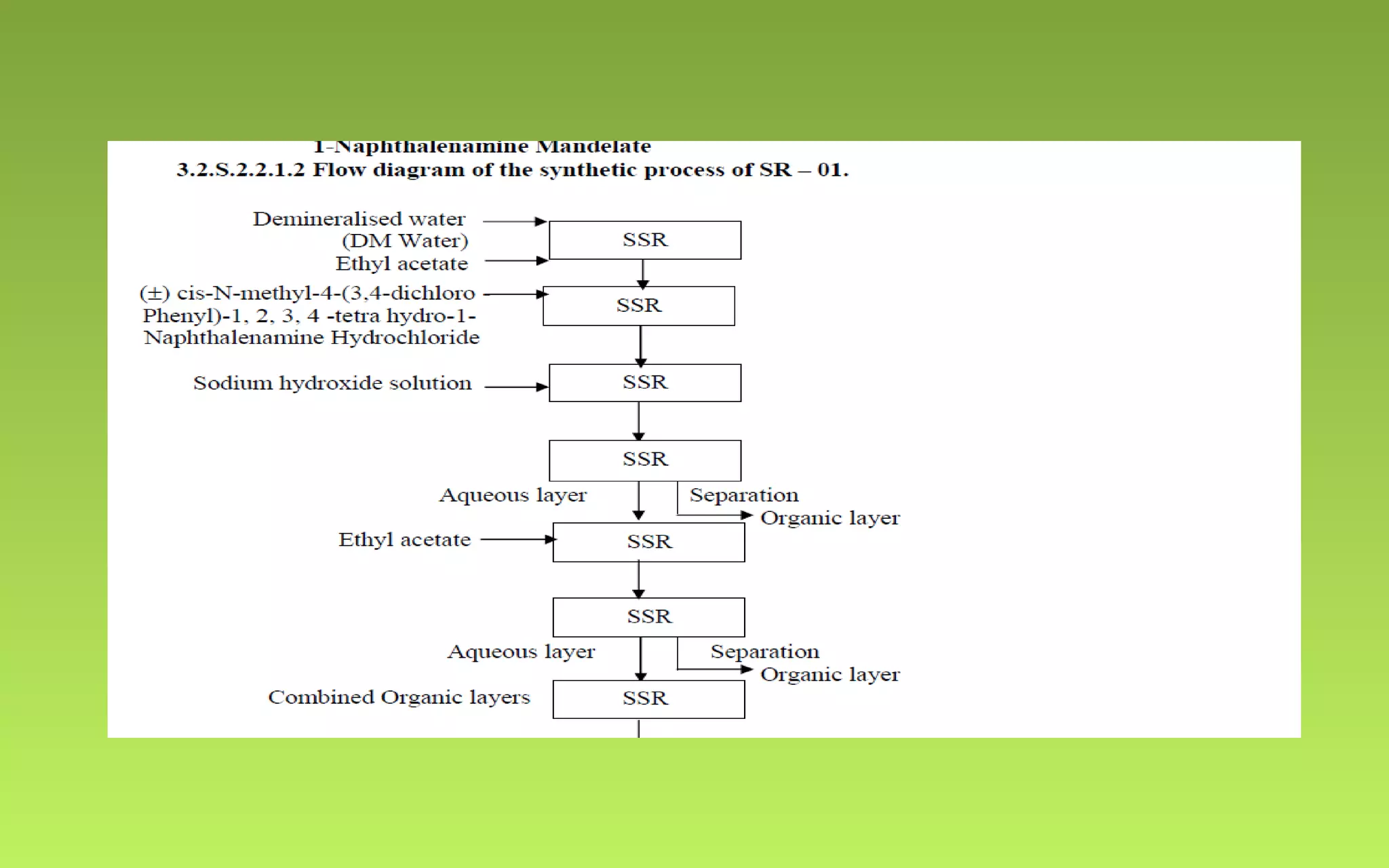




















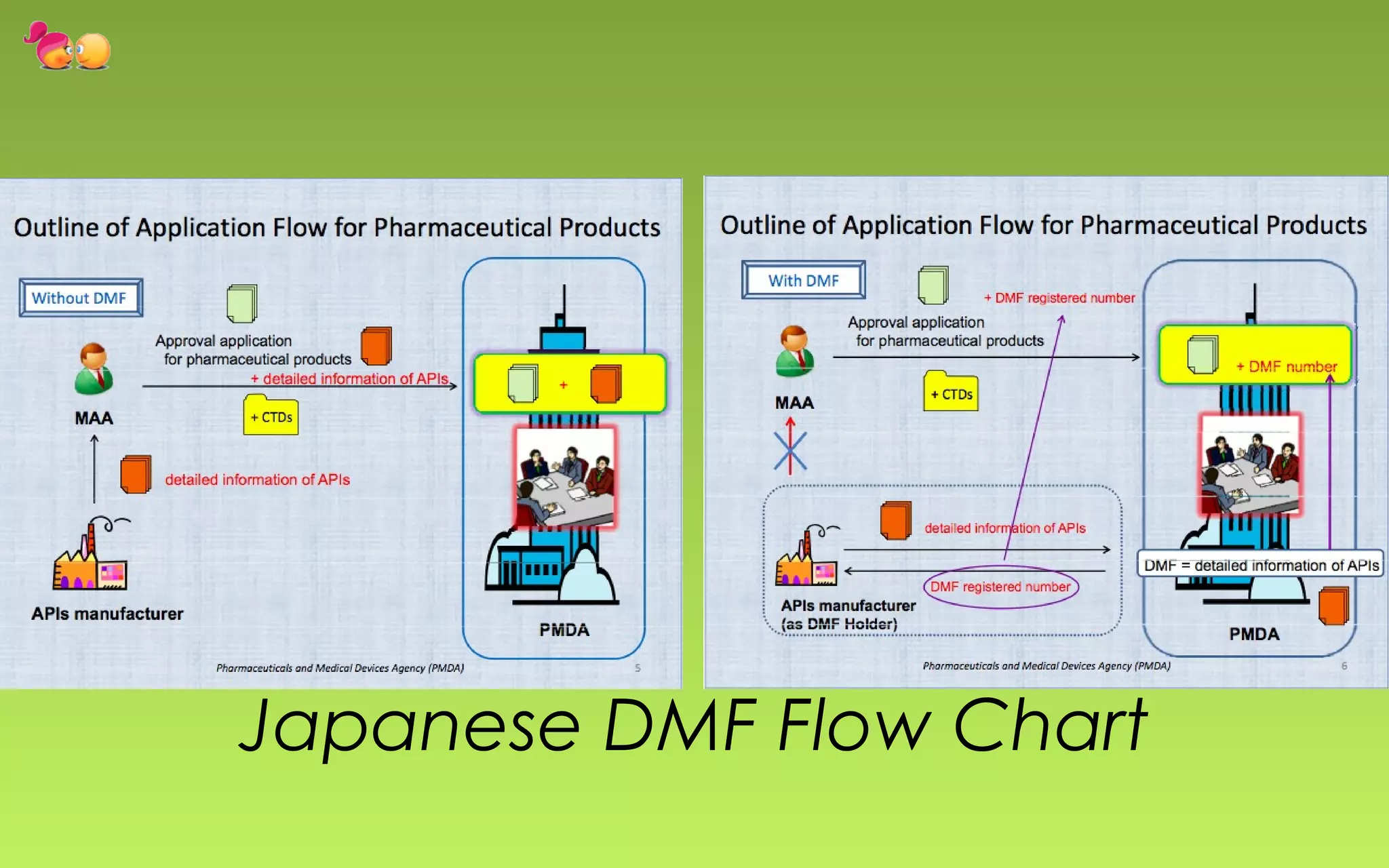








This document provides an overview of Drug Master Files (DMFs), including: - DMFs allow companies to provide confidential manufacturing information to regulatory agencies to support drug applications without publicly disclosing sensitive details. - DMFs are "closed" in the US, meaning only the regulatory agency reviews the file. In Europe, DMFs have both open and closed portions. - The document outlines the key differences between the US and European DMF systems, as well as providing details on the types of information included in a DMF and the processes for submitting and reviewing DMFs with regulatory agencies.











































































































